
































Chapitres
- Présentation de Michel Caboche02'32"
- Introduction05'16"
- Séquençage et génome des plantes21'20"
- Pourquoi la polyhaploïdie chez les plantes11'59"
- Du gène au phenotype15'39"
- Conclusion00'52"
- Questions18'11"
Notice
La génomique végétale et les plantes cultivées - Michel Caboche
- document 1 document 2 document 3
- niveau 1 niveau 2 niveau 3
Descriptif
Une conférence du cycle : Qu'est ce que la vie ? Où en est la connaissance du génome ?
Par Michel Caboche, Directeur adjoint de l’unité de Recherche en Génomique Végétale INRA – CNRS – Université d’Evry

Documentation
Documents pédagogiques
La génomique végétale
M.Caboche – Unité de recherches en génomique végétale
INRA – CNRS – UEVE, Evry
Introduction
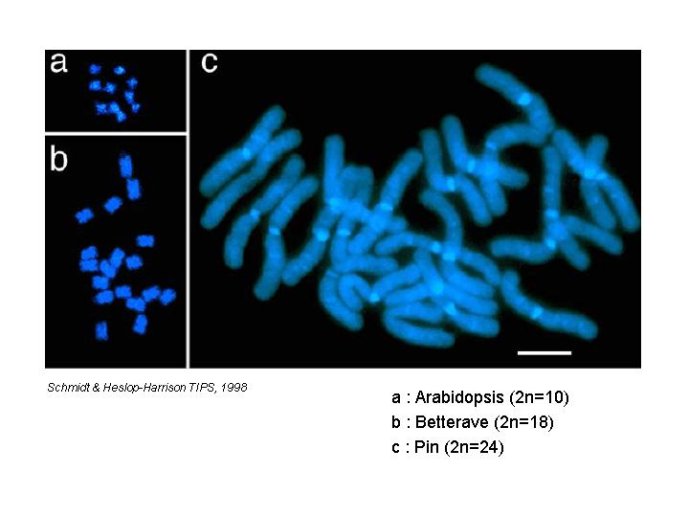
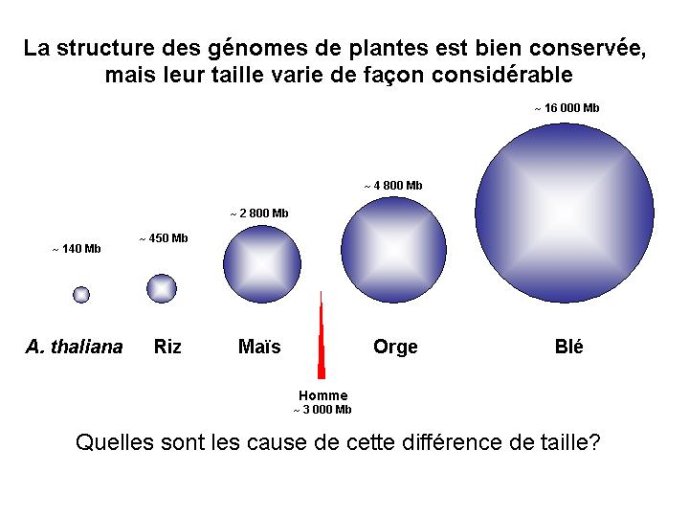
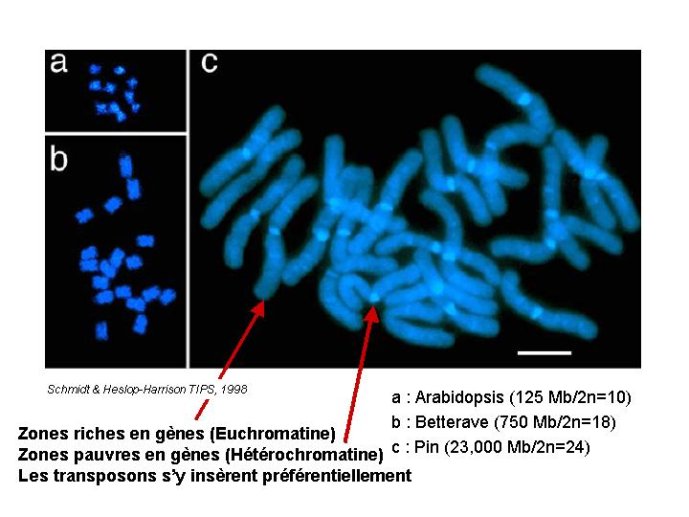
La génomique est une branche de la biologie qui porte sur l’étude des génomes, qui constituent le support moléculaire des caractères héréditaires des êtres vivants. L’étude de ces caractères héréditaires a été initiée par Gregor Mendel sur une plante, le petit pois. Ces travaux ont montré que les caractères héréditaires sont transmis en descendance selon des lois statistiques (lois de Mendel. Ces lois , oubliées puis redécouvertes par Morgan et ses élèves étudiant la mouche drosophile se sont révélées valables aussi bien pour les plantes que les animaux. De plus, les travaux de Morgan ont montré que ces caractères héréditaires appelés gènes sont disposés comme un chapelet de perles sur les chromosomes. Les lois de Mendel permettent de mesurer les distances qui séparent ces caractères héréditaires sur les chromosomes et de proche en proche, en analysant un grand nombre de gènes, de constituer une carte génétique de l’espèce étudiée. Les chromosomes, supports de ces caractères, sont constitués d’un fil d’ADN « emballé » dans une matrice protéique, la chromatine. Les travaux des biologistes moléculaires ont démontré que l’ADN est le support moléculaire des gènes qui sont constitués d’un enchaînement de quatre molécules appelées bases (ATGC). Les techniques de visualisation des chromosomes montrent que leur structure n’est pas homogène. Certaines zones sont très compactes et accumulent les colorants (hétérochromatine) et d’autres zones à coloration régulière sont appelées euchromatiques. On observe aussi une grande hétérogénéité de taille des chromosomes eux-mêmes, selon les espèces (Figure 1). Cette différence reflète-t-elle une différence dans le nombre des gènes présents dans différentes espèces ? La génomique a permis d’élucider cette question et d’expliquer la base de ces hétérogénéités.
Génomique structurale
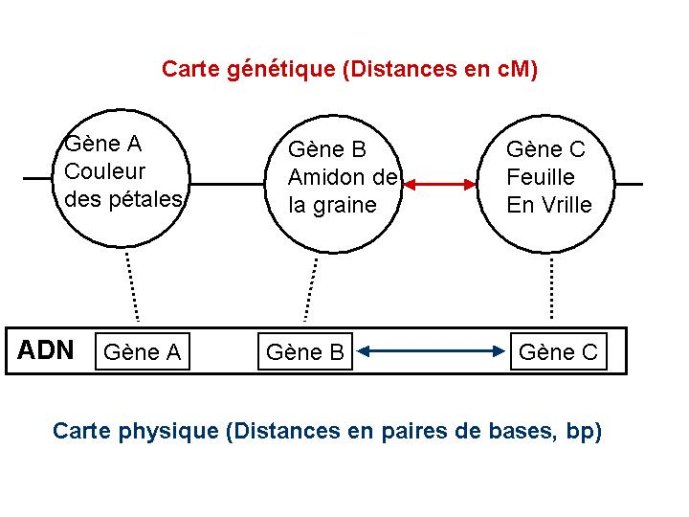
Comment se relie la carte génétique aux fils d’ADN présents dans les chromosomes ? Les techniques de génétique et de biologie moléculaire ont permis de localiser avec une grande précision la distance séparant deux gènes à la fois sur la carte génétique (distances mesurées en centi-Morgans, obtenues par analyse génétique) et sur le fil d’ADN (distances mesurées en nombre de bases sur l’ADN, obtenues en clonant un fragment d’ADN portant les deux gènes étudiés et en analysant sa séquence, Figure 2).
Le séquençage des génomes de plantes
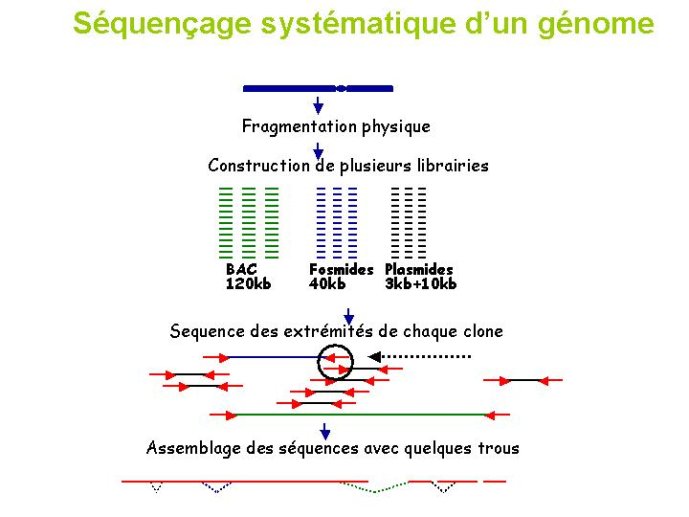
L’analyse de l’ADN constituant les chromosomes a fait des progrès gigantesques au cours de la dernière décennie. Les techniques de séquençage ont permis de passer de l’analyse d’un gène et de son proche voisinage, à l’analyse d’un génome complet. La taille moyenne d’un gène de plante est de l’ordre de 5000 bases, celle d’un génome de plante de petite taille est de 100 000 000 bases (100 Mb), soit 20000 fois plus. La stratégie de séquençage d’un génome complet est illustrée dans la figure 3. Elle consiste a casser le génome étudié en petits fragments, à isoler (cloner) ces fragments et à les séquencer afin de déterminer l’enchaînement des bases qui constituent cet ADN. Les outils informatiques permettent de comparer leurs séquences des fragments du génome étudié et du fait des chevauchements de séquence entre fragments de reconstituer la séquence complète d’un génome. Le premier génome de plante a été séquencé en l’an 2000. La petite crucifère Arabidopsis thaliana a été choisie du fait de la taille relativement petite de son génome (140 M bases) et de son utilisation aisée pour les travaux de génétique (Figure 4). D’autres génomes de plante sont maintenant séquencés (ex : Riz, peuplier, vigne) ou en cours de séquençage (luzerne diploïde, tomate, soja, maïs, etc…).
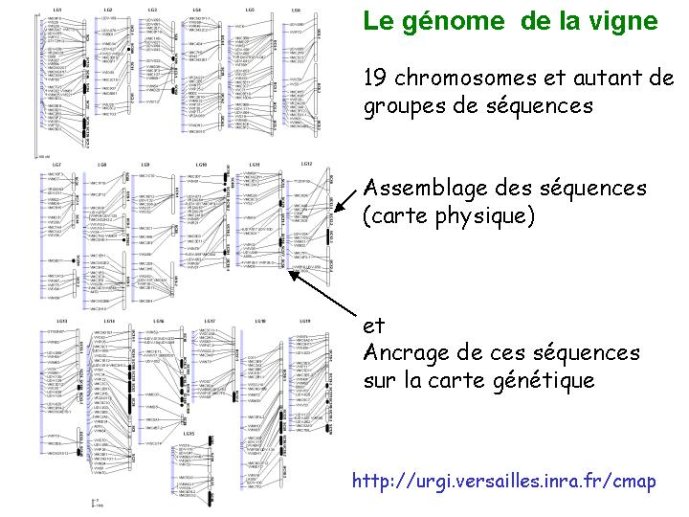
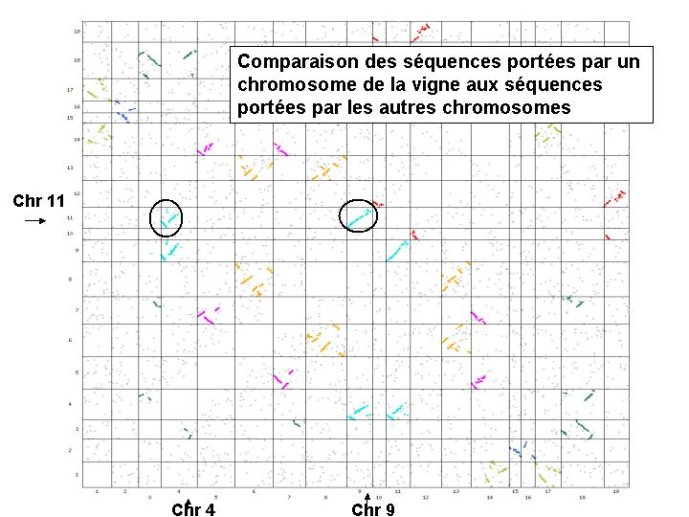
Les gènes de l’espèce séquencée qui sont déjà cartographiés et identifiés au niveau moléculaire permettent d’ancrer les cartes génétiques sur les séquences de l’ADN des chromosomes. Un exemple en est donné pour le génome de la vigne récemment séquencé par un consortium Franco-Italien (Figure 5). On observe que les gènes se suivent dans le même ordre sur la carte génétique et sur l’ADN des chromosomes , mais les distances entre gènes ne sont pas proportionnelles sur les deux cartes. Ceci est dû au fait que la recombinaison entre chromosomes au cours de la reproduction sexuée est réduite en certains endroits, en particulier au niveau des centromères des chromosomes.
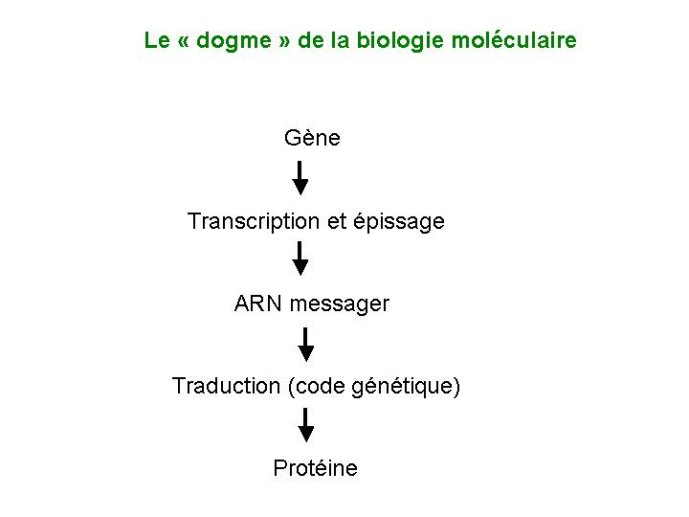
L’annotation des génomes
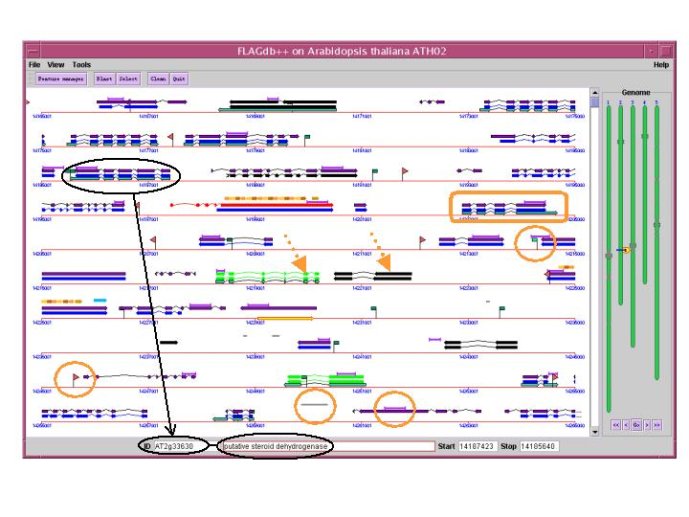
L’identification de la séquence d’un génome est un premier pas décisif pour son analyse. Il faut ensuite « lire » cette séquence, afin d’y déceler la présence des gènes qui s’y trouvent. Ce travail est intitulé « annotation ». Le principe de cette analyse repose sur le « dogme » de la biologie moléculaire. A tout gène correspond un transcrit constitué d’ARN, lui même utilisé par les ribosomes pour fabriquer une protéine. La correspondance entre séquence du gène (enchaînement de bases) et séquence de la protéine (enchaînement d’acides aminés) repose sur le code génétique. : à la succession trois bases appelée codon correspond un acide aminé particulier, à l’exception de trois codons dévolus à l’interruption/terminaison de la séquence protéique. Les outils bioinformatiques, en faisant correspondre à une séquence d’ADN toutes les séquences protéiques pour lesquelles elle peut coder potentiellement, permettent de localiser la présence possible d’un gène. Dans la pratique l’annotation d’un génome comporte trois étapes : une première étape où sont identifiées les séquences répétées présentes dans le génome, afin de les masquer. En effet ces séquences répétées sont occasionnées par la présence de transposons, éléments génétiques qui ne sont pas assimilés à des gènes, et constituent une sorte de bruit de fond dans l’analyse des génomes. Une seconde étape localise des séquences codantes potentielles en utilisant les règles du code génétique. Une troisième étape consiste à comparer la séquence étudiée à d’autres séquences déjà répertoriées comme présentes dans un gène déjà connu. Plus la séquence du gène étudié « ressemble » a celle d’un gène déjà connu, et plus ces deux gènes ont une fonction proche voire identique.. De cette manière l’annotation d’un génome permet de lister les gènes présents dans ce génome. Pour Arabidopsis ce nombre est d’environ 25000 gènes. Il faut garder en tête que cette annotation repose de manière ultime sur des outils d’analyse statistique qui prédisent la probabilité de présence d’un gène et non sa présence certaine.. D’autres techniques dites d’analyse fonctionnelle permettront de valider ou non cette prédiction, mais ces autres méthodes sont beaucoup plus lourdes à mettre en place que l’annotation bioinformatique qui peut être menée à bien en quelques semaines. Un exemple d’annotation de séquence est présenté en figure 6. Les différentes couleurs employées prédisent chacune des propriétés des gènes identifiés (ex : localisation intracellulaire de la protéine codée par le gène). On notera la structure morcelée des séquences codantes des gènes, due à la présence d’introns dans les gènes d’espèces eucaryotes dont les plantes font partie. Un bilan des gènes identifiés dans le génome d’Arabidopsis et du riz est présenté en Figure7. Noter la présence de gènes d’un type nouveau, des gènes qui ne codent pour aucune protéine, mais qui génèrent de petits ARN non traduits en protéines, petits ARN dont le rôle dans les mécanismes de régulation a été découvert au cours de la dernière décennie.
La structure des génomes
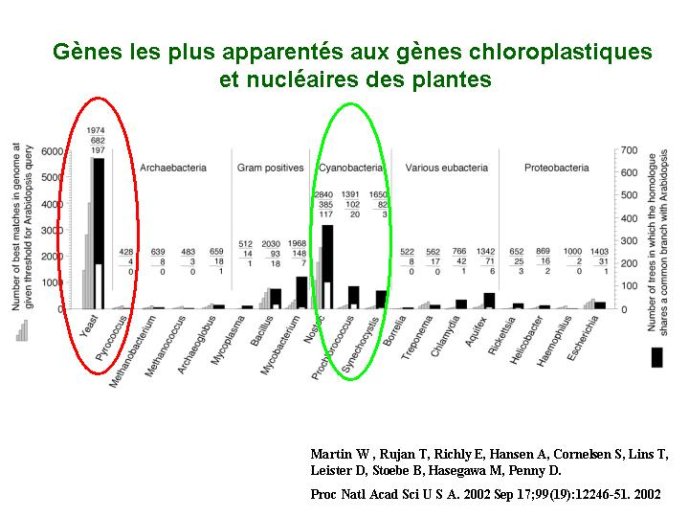
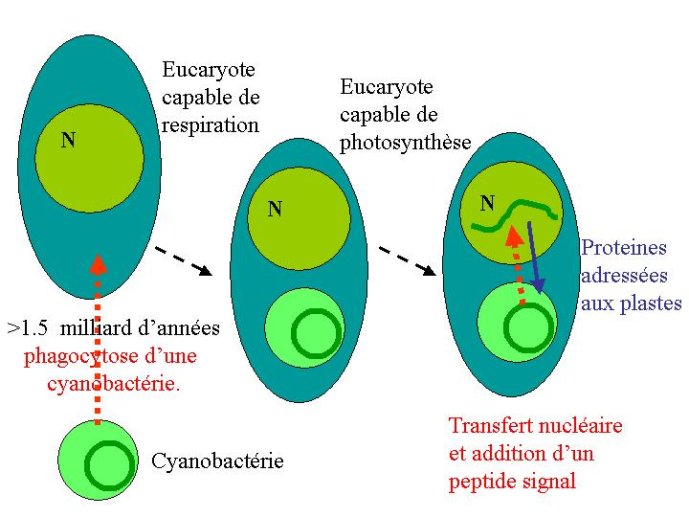
L’accès à la séquence d’un génome ouvre de multiples possibilités d’analyse. La comparaison des gènes présents dans les génomes des plantes avec les gènes d’autres espèces confirme l’origine symbiotique des plantes. Cette comparaison montre qu’un grand nombre de gènes de plantes ressemblent à des gènes de levure ou de mammifères (figure 8). Ceci s’explique simplement par le fait que levures, mammifères et plantes sont des eucaryotes issus probablement d’un ancêtre commun unicellulaire. Plus surprenante est l’observation de similitudes importantes entre de nombreux gènes de plantes et des gènes de bactéries photosynthétiques (Figure 8). Ceci s’explique par une hypothèse : les algues et plantes photosynthétiques seraient issues de la symbiose d’une cellule eucaryote non photosynthétique avec une cyanobactérie dont les gènes sont passés, au cours de l’évolution, de la cyanobactérie devenue chloroplaste au noyau de la cellule hôte.
Les génomes de plantes sont polyploïdes
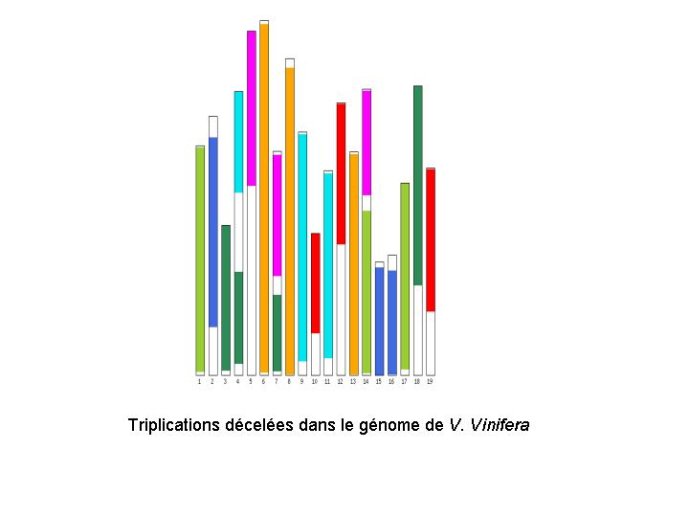
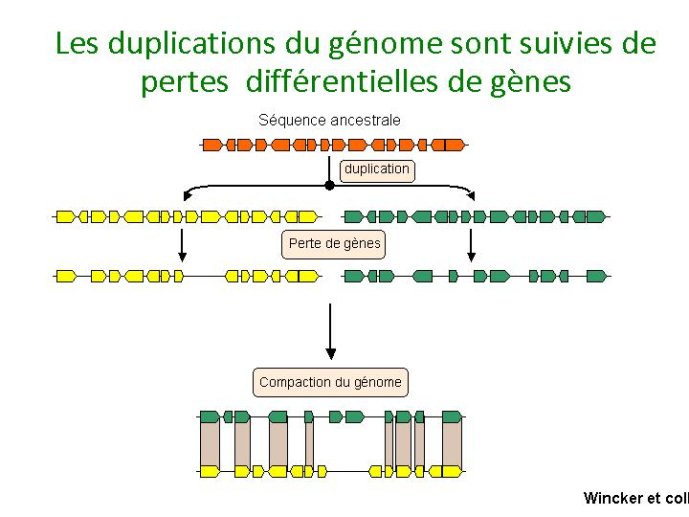
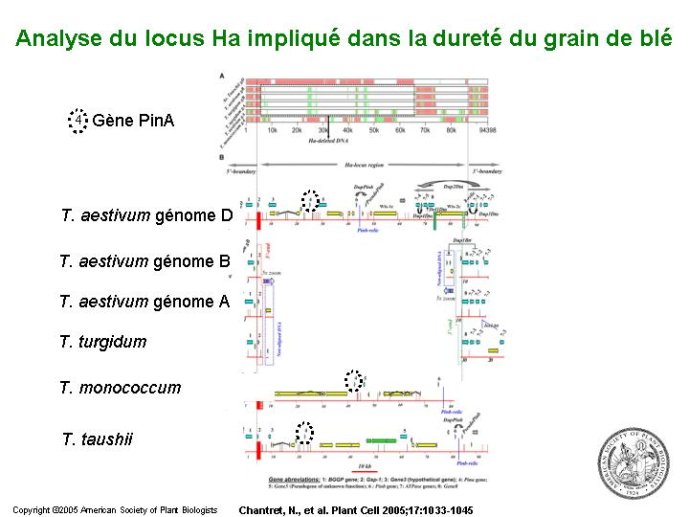
La comparaison des cartes génétiques d’espèces végétales apparentées (ex : céréales) a montré une conservation de la succession des gènes le long des chromosomes (Figure 9). L’analyse des séquences des chromosomes a amplement confirmé ces observations et montré que l’ordre des gènes sur les chromosomes est d’autant mieux conservé que les espèces sont phylogénétiquement apparentées. La comparaison des séquences présentes dans un même génome a occasionné quelques surprises. Dans de nombreuses espèces on constate que des fragments de chromosomes présentent des homologies de séquence avec d’autres fragments chromosomiques, l’ordre d’une partie des gènes étant conservée. Ceci est interprété comme un événement de duplication d’une partie du génome ancestral à l’espèce étudiée, ou une duplication complète de ce génome ancestral, ou enfin une addition de deux génomes apparentées. Ainsi la trace d’un événement de triplication a été décelée chez la vigne, mais aussi chez le peuplier et l’arabette. Dans ces deux dernières espèces d’autres évènements de duplication de leur génome viennent complexifier leur structure. Ces évènements de duplication sont parfois extrêmement récents comme l’illustrent l’exemple du colza issu d’une addition récente des génomes du chou et de la navette et l’exemple du blé tendre issu de l’addition de trois génomes A, B et D (Figure 10). Pourquoi ces phénomènes de polyploïdisation sont-ils fréquents chez les plantes, plus que dans le règne animal ? Il n’y a pas d’explication claire à cette question. L’addition de deux génome s’apparente à la production d’hybrides dont la vigueur dépasse celle des plantes homozygotes. Les espèces polyploïdes sont fréquemment plus vigoureuses que leur progéniteurs, elles ont donc un avantage sélectif dans l’environnement, et un intérêt direct pour l’agriculteur qui cherche à augmenter ses rendements. Si les évènements de polyploïdisation des génomes végétaux sont fréquents, on s’attend à ce que le nombre des gènes soit très variable d’une espèce à l’autre. Or un génome comme celui d’Arabidopsis ou de la vigne comportent chacun moins de 30 000 gènes. Superposé aux processus de polyploïdisation, un processus de perte de gènes dupliqués est observé. Lorsque deux copies d’un génome ancestral co-existent dans un même génome on constate que des pertes de gènes sont observées sur l’une ou l’autre des deux copies du génome ancestral, mais pas sur les deux à la fois. Ceci est illustré dans la figure11. Un exemple saisissant du processus de perte de gènes concerne le gène de dureté du grain PinA, présent dans les trois génomes des espèces ancestrales du blé, absent des génomes A et B mais présent seulement dans le génome D du blé tendre, lui conférant ainsi son caractère panifiable. Simultanément on observe que des gènes conservés en double copie vont souvent acquérir des spécificités divergentes (ex : expression dans des tissus différents). Ce mécanisme de polyploïdisation/perte de gène/différenciation est à l’œuvre et aboutit à une création de biodiversité.
Les éléments transposables et la taille des génomes
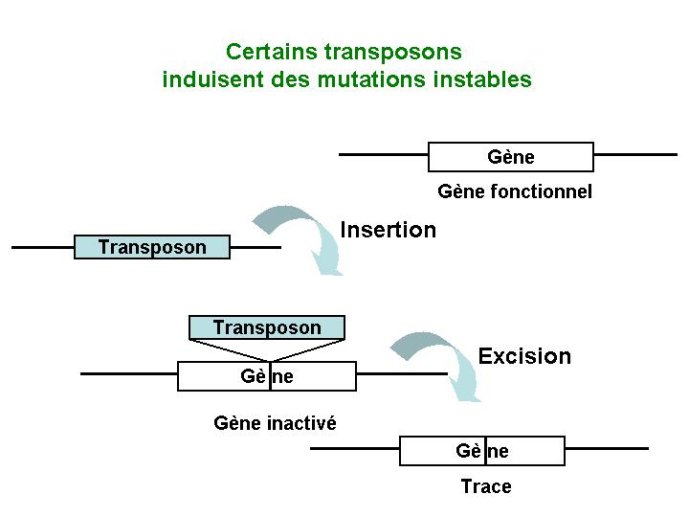
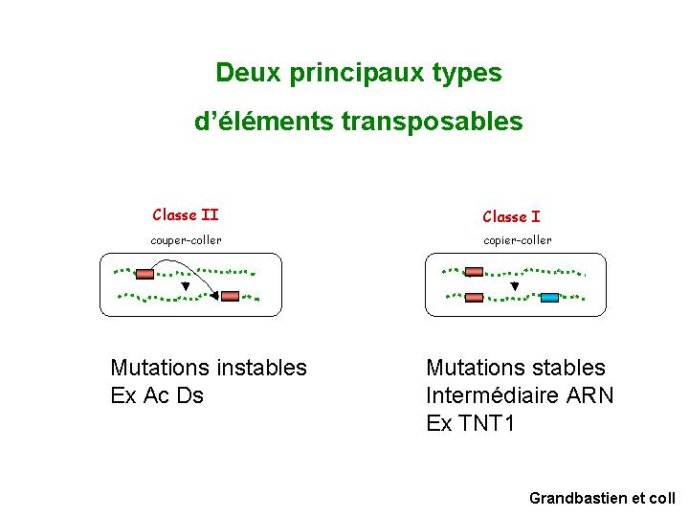
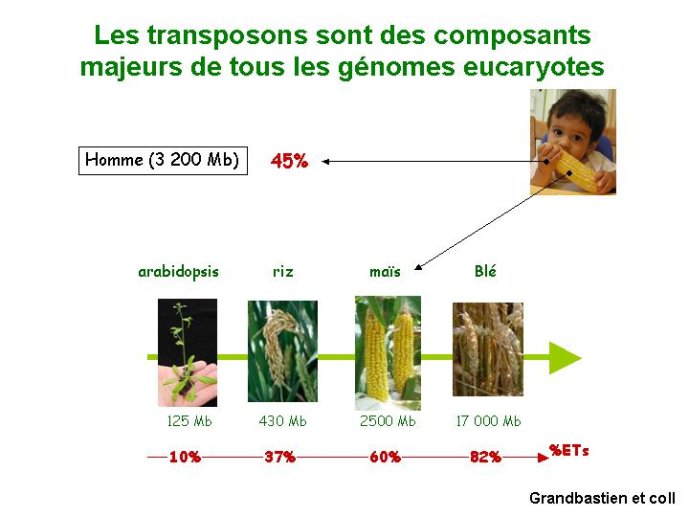
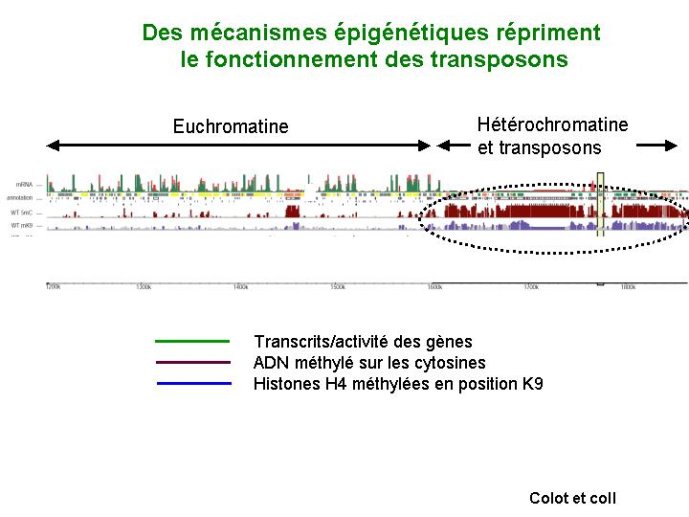
Un autre élément important dans la création de biodiversité chez les plantes réside dans la présence d’éléments transposables dans leur génome. Présents quelques fois en très grand nombre, ces éléments transposables ont été identifiés dans les génomes de nombreuses espèces. Cette présence plus ou moins abondante explique pour l’essentiel les différences de taille observées entre génomes végétaux. Les éléments transposables ont été initialement identifiés chez le maïs par B. Mac Clintosch. Ce sont des fragments d’ADN qui bougent dans le génome, et ce faisant provoquent des mutations. Il en existe deux grandes classes. Les éléments de classe I génèrent des copies surnuméraires qui s’accumulent dans les génomes et provoquent une augmentation de leur taille. Les éléments de classe II s’excisent du site où ils sont insérés pour s’insérer ailleurs dans le génome, provoquant ainsi des mutations instables (Figure 12). Les éléments transposables représentent moins de 10 % des séquences du génome d’Arabidopsis, mais 82 % du génome du blé (Figure 13). Induisant des mutations et des modifications des séquences dans lesquelles ils s’insèrent, les éléments transposables sont de puissants facteurs de génération de variabilité génétique. Les éléments transposables s’accumulent préférentiellement dans les zones hétérochromatiques des chromosomes. Cette localisation est associée à leur inactivation par des processus épigénétiques. On imagine que si une famille de transposons venait à être active en permanence, elle envahirait littéralement le génome, induisant de multiples mutations létales. Leur inactivation est donc un processus primordial mettant en jeu des processus de methylation de l’ADN et de modification des histones.
Génomique fonctionnelle
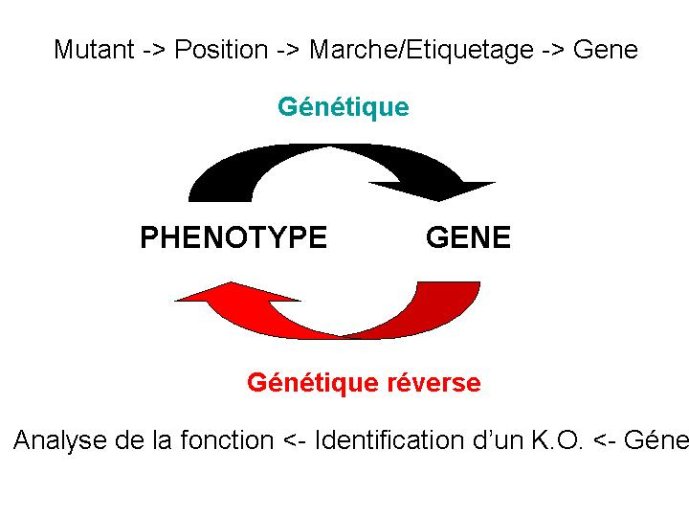
Les travaux d’annotation constituent un premier pas dans l’identification de la fonction des gènes présents dans le génome d’une espèce végétale. D’autres outils sont nécessaires à l’identification précise de la fonction de ces gènes. Clonage positionnel
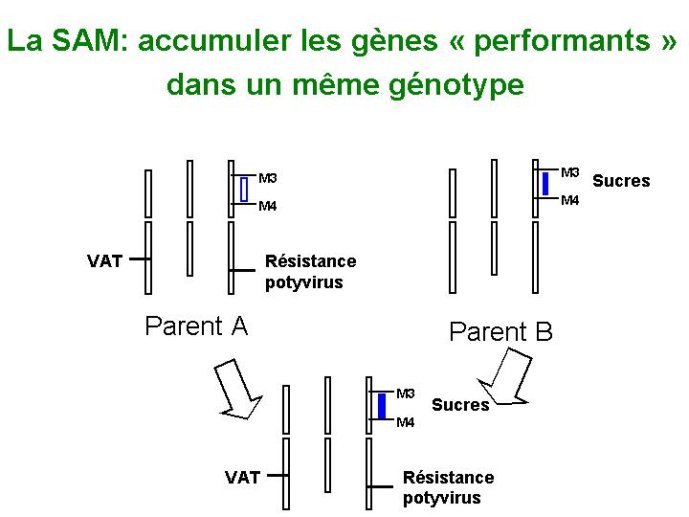
Chez une espèce comme Arabidopsis pourtant intensément étudiée, moins de 50% des gènes ont une fonction connue. Une approche universelle permettant l’identification du support moléculaire d’un caractère héréditaire est le clonage positionnel. Cette approche repose sur une cartographie extrêmement fine du gène recherché par rapport à des marqueurs moléculaires situés à proximité de ce gène. Ces marqueurs moléculaires jouent le rôle de bornes kilométriques pour localiser le gène dans la séquence ADN. C’est cette méthode que nous avons employé à l’URGV pour localiser le gène VAT conférant la résistance aux pucerons chez le melon. Un génotype résistant et un génotype sensible (Figure 14) ont été croisés, et l’étude de la ségrégation en descendance du locus de résistance a permis de construire une carte de l’ADN à proximité de VAT (Figure 15). C’est un gène de type NBS LRR qui s’est révélé être le gène VAT. Il appartient à la grande famille des gènes de résistance aux pathogènes. Cette démarche est particulièrement bien adaptée à l’identification de gènes supports de caractères agronomiques dans des espèces cultivées dont le génome n’a pas encore été séquencé. Une fois ces gènes identifiés, il devient relativement aisé de les associer dans un même génotype pour constituer une variété élite cumulant par exemple des gènes de résistance à des pathogènes, et par ailleurs des gènes contrôlant la teneur des fruits en sucre. Cette démarche est appelée sélection assistée par marqueurs (Figure 16).
L’étiquetage de gènes
Le clonage positionnel est une méthode lourde, et peu adaptée aux techniques dites à haut débit. Analyser la fonction d’un grand nombre de gènes nécessite d’autres outils. Une seconde approche consiste à générer une collection de mutants par insertion au hasard dans le génome d’une séquence ADN connue. C’est une méthode que nous avons développé à l’INRA, en tirant profit d’une technique de transformation à haut débit par inoculation d’Agrobacterium tumefasciens, une bactérie qui insère spontanément un fragment de son génome, appelé ADN-T dans les cellules de plante. Une collection de 50 000 lignées d’Arabidopsis porteuses chacune d’un fragment ADN-T connu dans leur génome a été produite (Figure 17). Cette collection a été criblée pour la présence de mutants affectés dans diverses fonctions (système reproducteur, remplissage de la graine, attaque des pathogènes, adaptation aux stress hydriques, etc…). Lorsqu’un mutant de la collection est affecté dans l’une de ces fonctions, il est aisé d’identifier le gène cible de la mutation qui est « étiqueté » par l’insertion d’ADN-T
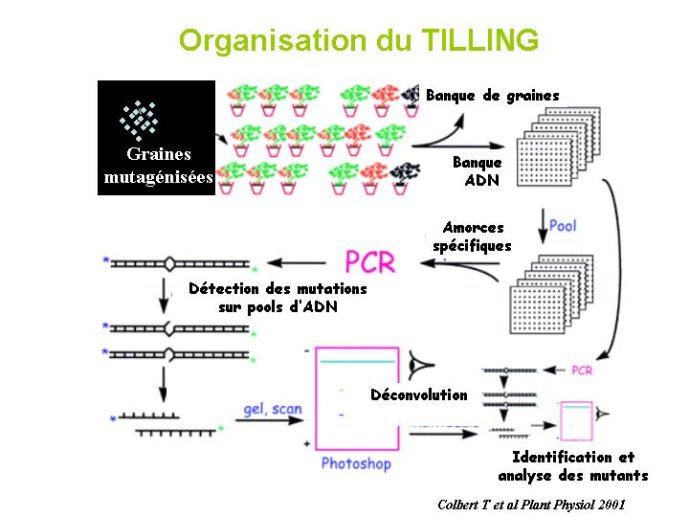
Le TILLING
Une approche de génétique reverse (allant de la séquence du gène à sa fonction) particulièrement bien adaptée à l’étude de la fonction des gènes chez les plantes cultivées concerne la technique de TILLING. Le principe est simple : on induit des mutations dans le génome d’une plante homozygote par mutagénèse chimique sur ses graines. En seconde génération les familles issues de mutagénèse sont analysées pour la présence de mutations affectant le gène d’intérêt étudié. Au lieu d’effectuer un séquençage du gène dans plusieurs plantes de chaque famille, ce qui serait lourd et onéreux, on hybride l’ADN de chaque famille à un ADN témoin non muté. S’il y a une différence de séquence entre l’ADN témoin et l’ADN testé,, l’ADN hybride obtenu comportera un mis-appariement qui sera décelé par une méthode biochimique très sensible (Figure 17). Cette technique permet d’obtenir assez facilement une collection de mutations affectant un gène particulier. Les plantes identifiées comme mutantes sont ensuite étudiées pour leur phénotype. L’intérêt de la technique est double. Elle permet de manipuler une population non OGM, qui ne nécessite aucun confinement en serre. De plus l’efficacité de la mutagénèse est indépendante de la taille du génome étudié. Nous avons ainsi développé des programmes de TILLING chez le pois à la fois récalcitrant à la transformation par Agrobacterium, et pourvu d’un génome de très grande taille.
L’analyse du transcriptome
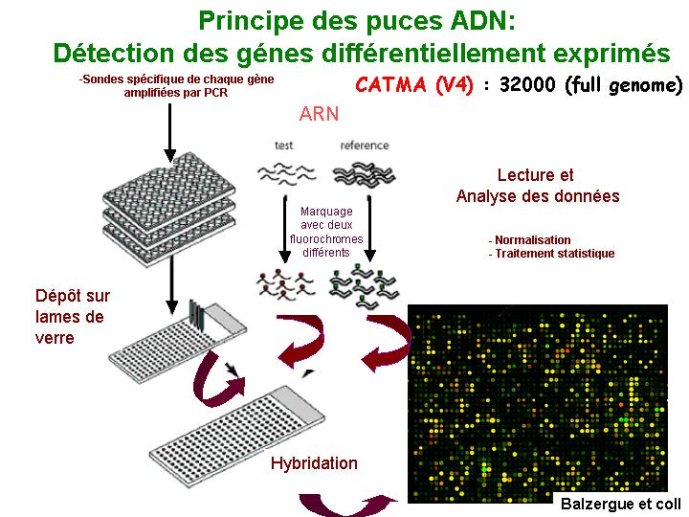
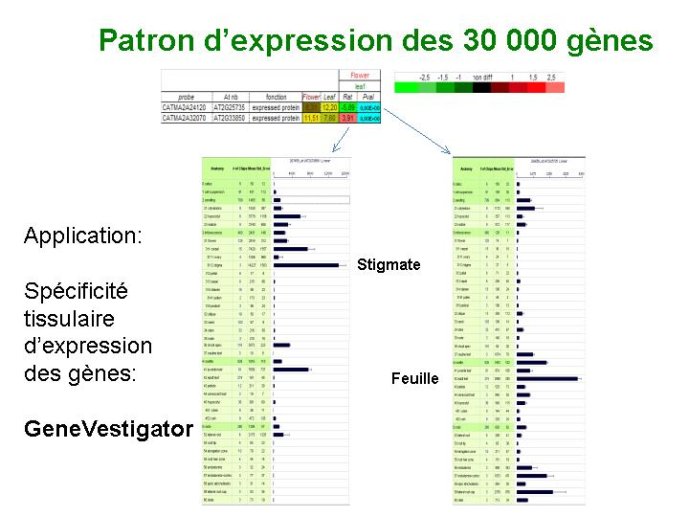
Une quatrième approche permettant d’identifier la fonction d’un gène consiste à analyser ses caractéristiques d’expression. Ceci se fait traditionnellement par emploi d’une technique de gène « rapporteur » qui nécessite la production de plantes transgéniques. Cette méthode a l’inconvénient d’être « à bas débit ». Une alternative, dite d’analyse du transcriptome repose sur l’emploi de puces à ADN. Les 25 000 gènes d’Arabidopsis sont déposés individuellement sur un support de verre (la puce à ADN). Les lames sont alors hybridées avec des préparations de copies ADN des transcrits produits par les différents tissus de la plante. Ces copies ADN, marquées avec un fluorochrome vont s’hybrider avec les gènes déposés sur les lames. Un appareil de lecture va mesurer l’intensité des signaux d’hybridation de chaque gène, le signal étant d’autant plus important que le gène est activement exprimé (Figure 19). Le travail mené sur divers tissus, et dans diverses conditions expérimentales permet de dresser un « portrait robot » des caractéristiques d’expression de chacun des 25000 gènes du génome d’Arabidopsis. Ces portraits robot sont ensuite comparés les uns aux autres et regroupés en classes fonctionnelles. Les membres de ces classes fonctionnelles qui ont une fonction connue permettent ainsi de faire des prédictions sur les fonctions des gènes encore inconnus dont les caractéristiques d’expression leur sont similaires.
Les méthodes d’analyse convergent pour identifier le rôle possible des gènes de fonction inconnue
Une cinquième approche permettant d’analyser la fonction d’un gène consiste à travailler au niveau de la protéine codée par ce gène. Cette protéine peut être localisée, par exemple par des techniques de fluorescence, dans le compartiment cellulaire ou elle joue son rôle. En parallèle il est possible de déterminer avec quelles autres protéines cette protéine interagit par les approches dites de double hybride, généralement conduites dans des levures. L’ensemble des données obtenues permet finalement d’étayer l’hypothèse faite sur la fonction du gène étudié. Analyse mutationnelle, analyse du transcriptome, identification des partenaires protéiques et localisation intracellulaire permettent de décrire la fonction du gène étudié. Dans notre laboratoire cette approche combinée est utilisée pour analyser la famille des PPR, famille multigénique quasi inexistante dans le règne animal dont le rôle est de contrôler la mise en place des fonctions chloroplastiques et mitochondriales d’expression des gènes (épissage des transcrits, éditions des séquences ARN, régulation de la transcription des gènes chloroplastiques ou mitochondriaux).
Conclusion
Ce rapide survol des approches génomiques utilisées pour étudier les génomes de plantes est incomplet. Chaque année de nouveaux outils d’analyse des génomes viennent compléter les approches (par exemple actuellement les outils d’analyse du protéome, les techniques de séquençage à très haut débit). Cet éventail de recherches, s’appuyant aussi sur les techniques d’analyse de la biodiversité des séquences nous apporte des informations sur l’évolution des génomes et sur les processus de domestication. La génomique apporte des outils nouveaux pour étudier la biodiversité des plantes, et pour constituer des ressources génétiques organisées et exploitables, une étape essentielle au travail d’amélioration génétique (Figure 20). Ces outils renouvellement en profondeur la démarche d’amélioration des plantes cultivées (introgression de caractères présents dans les espèces apparentées aux plantes cultivées, sélection assistée par marqueurs, génération de nouveaux allèles). La génomique végétale ouvre un champ de recherche et d’application immense dans lequel beaucoup de pays émergents investissent activement (Brésil, Inde, Chine, Mexique…). Souhaitons qu’il en soit de même dans un pays qui reste le troisième exportateur mondial dans le domaine semencier.
Bibiographie : pour en savoir plus…
Lurin C., Renou J.P., Bouchez D. La génomique et les outils d‘exploration fonctionnelle chez les plantes. Biofutur (2006) 265, 45-49.
Aubourg S, Delseny M, Lecharny A: L'organisation des génomes végétaux révélée par leur annotation.,Biofutur (2006) 265, 33-37.
Bouchez,D., Bendahmane, A., Lurin, C., Sturbois, B. Nouvelles approches génétiques. Biofutur (2006) 265,38-44
David, J., Loudet, O., Glaszmann, J.C. Le regard de la génomique sur la diversité naturelle des plantes cultivées.( 2006) Biofutur 266, 22-27
Jahier, J., Chalhoub, P., Charcosset, A. La domestication des plantes : de la cueillette à la post génomique. Biofutur (2006) 266, 28-33
Quetier,F. Salanoubat, M. Weissenbach, J.Le séquençage ds génomes nucléaires de plantes (2006) Biofutur 265,27-32.
Murigneux, A., Martinant, J.P., Barrière, Y. Apport de la génomique a l’amélioration des plantes. L’exemple du maïs fourrage. (2006) Biofutur 266, 34-40
J.F. morot-Gaudry et J.F. Briat. La génomique en biologie végétale INRA Editions (2004) 582 pages
Figures (N° power point)
Figure 1 Les chromosomes de trois espèces végétales.
Figure 2 Carte génétique et carte physique
Figure 3 Le séquençage génomique
Figure 4 Arabidopsis, espèce modèle
Figure 5 Colinéarité de la carte physique et de la carte génétique de la vigne
Figure 6 Annotation de 100kb de l’ADN génomique d’Arabidopsis.
Les gènes sont symbolisés par des traits de couleur épais. Les couleurs différentes correspondent à des prédictions basées sur des outils d’annotation différents. Ex : trait vert : la protéine correspondante est ciblée dans les chloroplastes
Figure 7 Inventaires des génes d’Arabidopsis et du riz.
Surprise ! il y a au moins autant de gènes dans un génome de plante que dans le génome humain.
Figure 8 Gènes apparentés aux gènes chloroplastiques et nucléaires des plantes. Ellipse rouge : gènes d’Arabidopsis homologues de gènes de levure
Ellipse verte : gènes d’Arabidopsis homologues de gènes de cyanobactéries
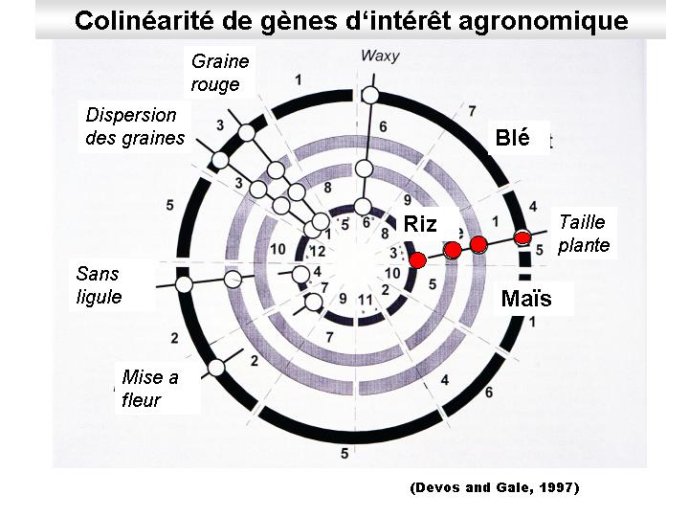
Figure 9 Les gènes d’importance agronomique (ex gènes de nanisme de la révolution verte) sont colinéaires dans les génomes d’espèces apparentées. L’exemple de trois céréales a été pris (blé , maïs, riz). Les chromosomes sont symbolisés par un cercle sur lequel ils sont mis bout à bout, les chiffres correspondant à la numérotation de ces chromosomes dans leurs espèces respectives. Les gènes impliqués dans une fonction commune se trouvent alignés sur un même rayon des cercles
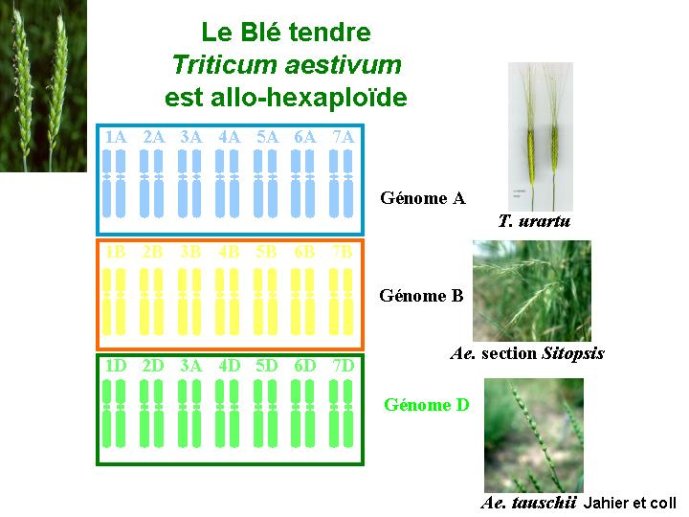
Figure 10
Le blé tendre, Tritcum aestivum est allo-hexaploïde.
Il est constitué de l’addition des génomes de trois céréales, Triticum urartu, Aegilops sitopsis et Aegilops Tauschii soit 3x7 chromosomes
Figure 11 Les duplications du génome sont suivies de pertes différentielles de gènes
Figure 12 Les transposons sont des composants majeurs de tous les génomes eucaryotes
Figure 13 Certains transposons induisent des mutations instables
Figure 14 Test de résistance au puceron Aphis gossypii dans des populations de melons en ségrégation
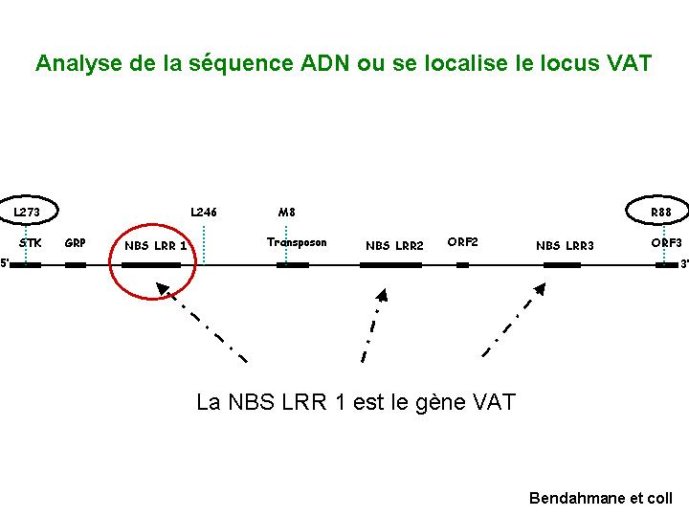
Figure 15 Analyse de la séquence ADN ou se localise le locus VAT.
La cartographie de VAT a permis de localiser ce gène entre deux marqueurs moléculaires L273 et R88. Un fragment d’ADN porteur de ces deux marqueurs a été identifié et séquencé. Plusieurs gènes candidats pouvant correspondre au gène VAT recherché ont été identifiés. Parmi eux le gène NBS LRR 1 s’est confirmé comme étant le gène VAT par cartographie fine.
Figure 16 Principe du TILLING
Figure 17 La sélection assistée par marqueurs (SAM) permet de cumuler les gènes « performants » dans un même génotype.
Ici trois gènes ont été cumulés : VAT, un gène de résistance à un virus et un gène contribuant à la teneur des fruits en sucres
Figure 18 La mutagénèse d’insertion nécessite de gérer beaucoup de plantes. 100000 familles de plantes d’Arabidopsis chacune porteuse d’une insertion ADN-T sont nécessaires à saturer le génome en insertions. Ici une serre permettant de gérer 500 familles. Du fait de leur taille ces collections sont le fruit de collaborations internationales.
Figure 19 Principe des puces à ADN : détection des gènes différentiellement exprimés dans deux conditions physiologiques différentes.
Figure 20 Biodiversité des fruits de piment
Dans la même collection
-
La génomique, nouvel observatoire des microbes de l'environnement
WeissenbachJeanUne conférence du cycle : Qu'est ce que la vie ? Où en est la connaissance du génome ? Par Jean Weissenbach, Directeur du centre national de séquençage, Génoscope d’Evry
-
Peut-on concevoir la cellule comme un ordinateur qui ferait des ordinateurs ? - Antoine Danchin
DanchinAntoineUne conférence du cycle : Qu'est ce que la vie ? Où en est la connaissance du génome ? Par Antoine Danchin, biologiste, directeur du département génomes et génétique à l’institut Pasteur
-
Pourquoi et comment faire des formes de vie nouvelles ?
MarlièrePhilippeUne conférence du cycle : Qu'est ce que la vie ? Où en est la connaissance du génome ? par Philippe Marlière
-
L'homme transgénique : des possibilités infinies - M. Radman, JC Weill
RadmanMiroslavWeillJean-ClaudeUne conférence du cycle : Qu'est ce que la vie ? Où en est la connaissance du génome ? Miroslav Radman, Généticien, Université Paris 5, faculté de médecine Necker Et Jean-Claude Weill,
-
Diversité du génome humain : de l'histoire des populations humaines aux maladies infectieuses
Quintana-MurciLluisUne conférence du cycle : Qu'est ce que la vie ? Où en est la connaissance du génome ? Par Luis Quintana Murci, Directeur de l’unité génétique évolution humaine, Institut Pasteur
-
Existe-t-il une génétique des comportements ? - Philip Gorwood
GorwoodPhilipUne conférence du cycle : Qu'est ce que la vie ? Où en est la connaissance du génome ? Par Philip Gorwood, Professeur en psychiatrie, Université Paris 7, Hôpital Louis Mourier (La
-
Génome et médecine prédictive - Jean-Louis Mandel
MandelJean-LouisUne conférence du cycle : Qu'est ce que la vie ? Où en est la connaissance du génome ? Par Jean-Louis Mandel, Médecin généticien, Collège de France
-
La carte d'identité génétique - Daniel Cohen
CohenDanielUne conférence du cycle : Qu'est ce que la vie ? Où en est la connaissance du génome ? Par Daniel Cohen, Généticien, fondateur du Généthon, PDG de PharNext
-
Génome et cancer - Mark Lathrop
LathropMarkUne conférence du cycle : Qu'est ce que la vie ? Où en est la connaissance du génome ? Par Mark Lathrop
-
Qu'attendre de la connaissance des génomes ?- A. Fagot-Largeault
Fagot-LargeaultAnneUne conférence du cycle : Qu'est ce que la vie ? Où en est la connaissance du génome ? Par Anne Fagot-Largeault, philosophe et psychiatre, Collège de France
-
Nouvelles questions et hypothèses sur l'origine et l'évolution des génomes - Patrick Forterre
ForterrePatrickUne conférence du cycle : Qu'est ce que la vie ? Où en est la connaissance du génome ? Par Patrick Forterre, Directeur du département de microbiologie de l’Institut Pasteur, professeur à l’Université
-
Algues marines, génomes et biotechnologies - Bernard Kloareg
KloaregBernardAlgues marines, génomes et biotechnologies par Bernard Kloareg. Principaux producteurs primaires en milieu océanique, les algues marines constituent une variété de lignées végétales qui










Sur le même thème
-
Les riboswitches, des solutions innovantes pour les biotechnologies et le diagnostic médical
BoudvillainMarcPrésentation en motion design du projet ANR HELISWITCH porté par Marc Boudvillain
-
Introduction à la randomisation mendélienne
BoisselMathildeIntervention de Mathilde Boissel lors de la journée consacrée à la randomisation mendélienne organisée par le groupe de travail "Statistique et Génomique" du RIS, le 27 octobre 2023
-
Complex traits and mendelian randomization : clues to clinical application
Gichohi-MainaJaredPrésentation d'une application de la randomisation mendélienne
-
PNLIPRP1, type 2 diabetes and cholesterol : applying mendelian randomisation to validate biologoica…
MaurinLucasPrésentation d'une application de la randomisation mendélienne
-
Etude de la structure tridimensionnelle du génome
VaroquauxNelleDétermination de la structure tridimensionnelle du génome
-
Dr Jianbin Wang - Genomes and transcriptomes of the parasitic nematode Ascaris
Nematodes of the genus Ascaris are important parasites of humans and swine, and related genera (Parascaris, Toxocara, and Baylisascaris) infect mammals of veterinary interest. Over the last decade,
-
Les vignes romaines : approches morphométrique et archéogénomique
BoubyLaurentLes vignes romaines : approches morphométrique et archéogénomique
-
Soirée de présentation de l'ouvrage "Sur la trace des suspects"
VaillyJoëlleChauvièreFabienneÀ l'occasion de la parution de l'ouvrage Sur la trace des suspects. L’incorporation de la preuve et de l’indice à l’ère de la génétique (coll. Le (bien) commun), les Éditions de la Maison des sciences
-
Le génome, sujet de l'évolution (cycle L'évolution récente de la biologie évolutive 1/4)
VeuilleMichelAprès que le mendélisme eut révolutionné la biologie évolutive après 1930, la nouvelle génétique évolutive a, à son tour, révolutionné le mendélisme et montré le rôle du génome et des forces qui
-
Daniel-Philippe de SUDRES : Comment les lieux neuro- modifient-ils nos affects?
Daniel-Philippe de SUDRES : Comment les lieux neuro- modifient-ils nos affects?
-
Le vieillissement, normal et pathologique (ASR n°15 - FHU)
Deux fédérations hospitalo-universitaires de l’Université de Lorraine combinent recherche expérimentale et recherche appliquée en centre hospitalier, avec l'ambition de mieux comprendre le
-
EEHU 2017 Génétique et médecine fœtale 03
Journée annuelle 2017 : Etre soignant à l’ère de la bioéthique Réflexions éthiques liées à la Génétique et à la Médecine Foetale La recherche sur l’embryon à l’ère de la génomique. Pierre











