L’informatique dans les sciences de la vie
Rechenmann
François
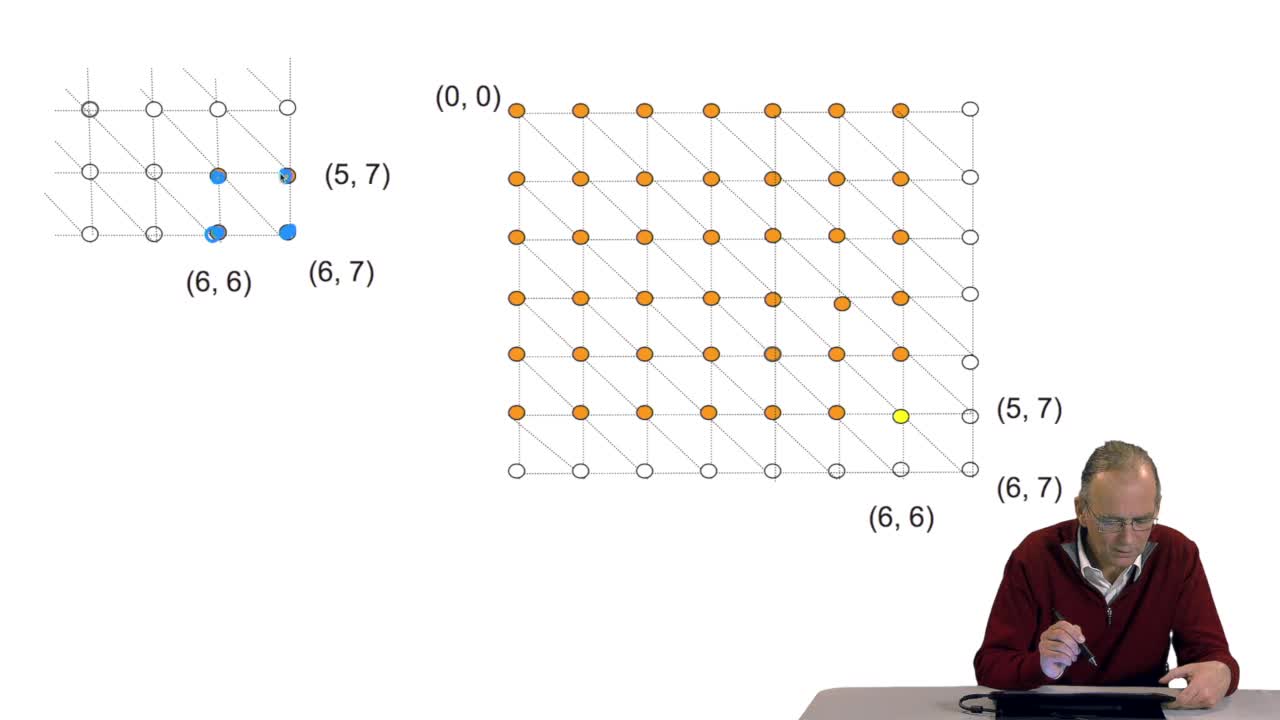
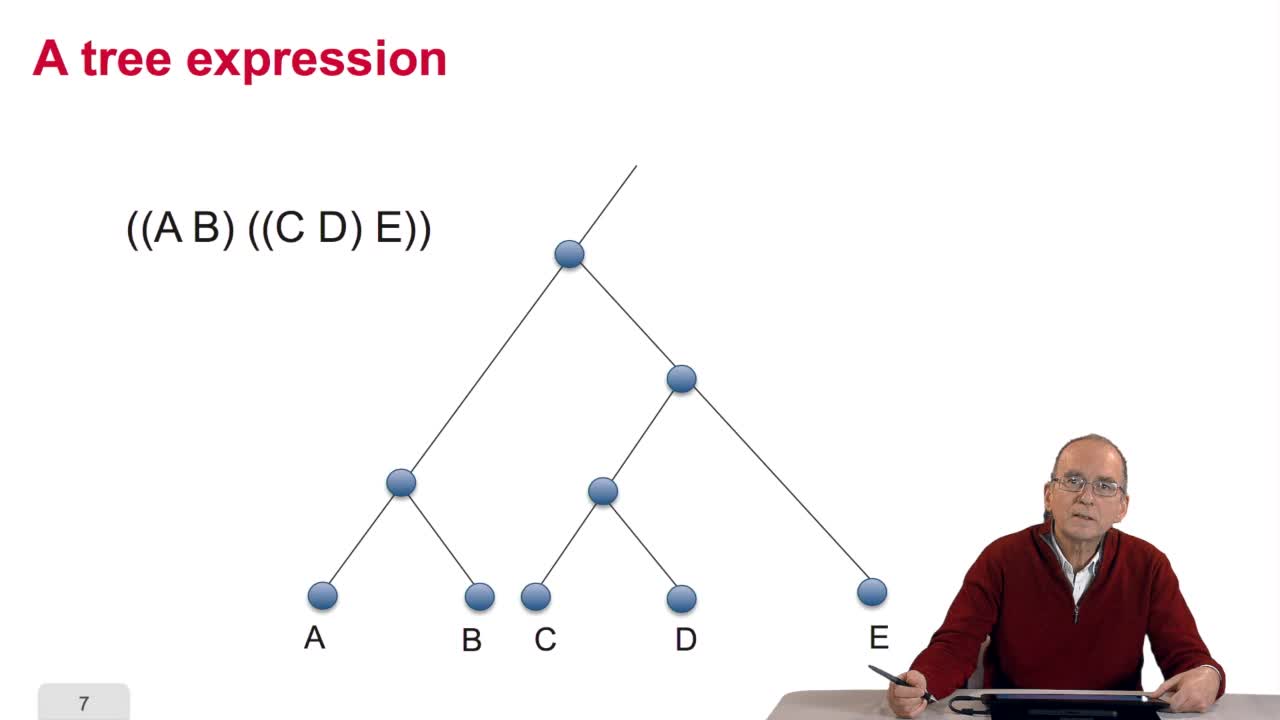
Dans cet exposé François Rechenmann propose un rapide survol des méthodes algorithmiques utilisées au niveau de l'analyse du génome. On y découvre que l'informatique est à la fois un outil