Notice
4.9. Recursion can be avoided: an iterative version
- document 1 document 2 document 3
- niveau 1 niveau 2 niveau 3
Descriptif
We have written a recursive function to compute the optimal path that is an optimal alignment between two sequences. Here all the examples I gave were onDNA sequences, four letter alphabet. OK. The writing of this recursive function is very elegant but unfortunately we will see now that it isnot very efficient in execution time. Let's see why. Remember the computing schema weapply during the recursion, for example here, to compute the cost of this node, we saw that it was required to computerecursively the cost of that node, that node and that node. OK but to compute the cost of that node here, you need to compute the cost of that one, that oneand that one again that is this cost which was computed in order to compute the code of the ending node here has to be recomputed in the recursive function to compute the cost of that node. In a more general way, to computethe cost of a node like this one, you need to compute all of these nodes here but to compute the cost of that node, you need to compute all the costs of these nodes again and again and again. So the cost of one node in thiswriting of the function is computed many, many, many times. It's because we use this recursive function so it was nice but it was expensive in terms of execution time. So we can imagine a new version of the algorithm which is not recursive but iterative in two phases. Let's see how it works.
Intervention / Responsable scientifique

Thème
Documentation
Dans la même collection
-
4.6. A path is optimal if all its sub-paths are optimal
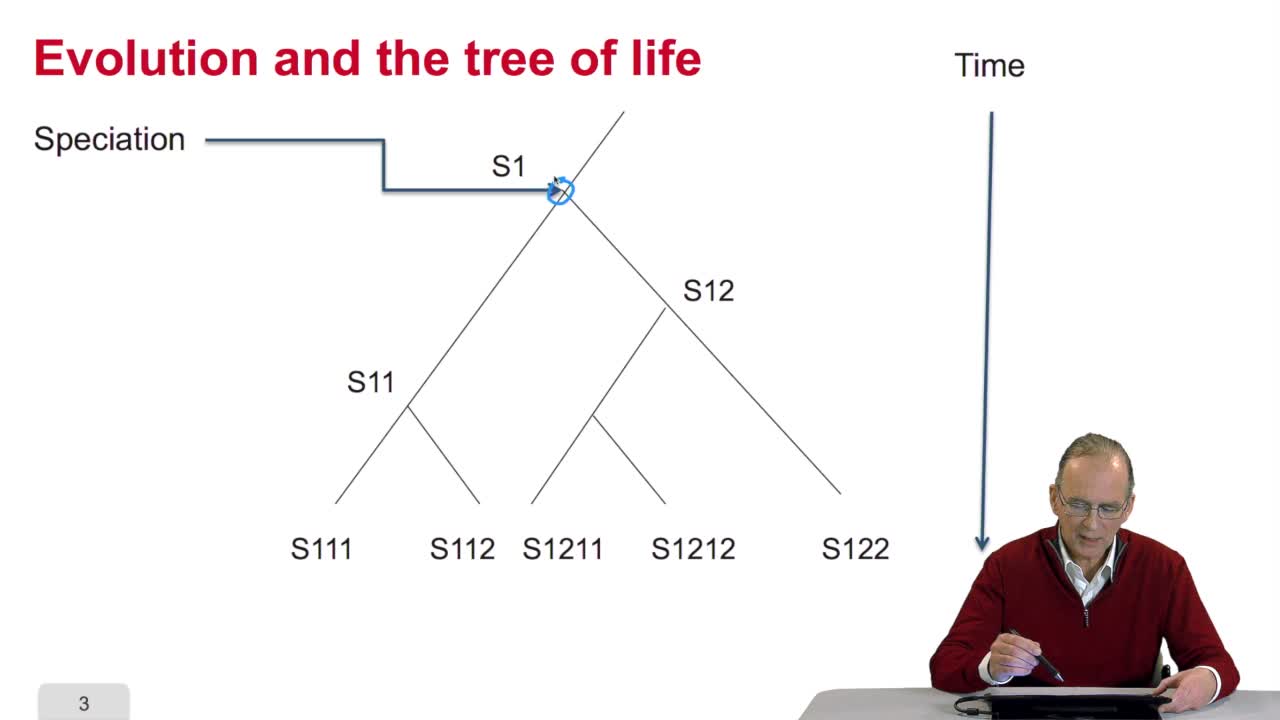
RechenmannFrançoisA sequence alignment between two sequences is a path in a grid. So that, an optimal sequence alignmentis an optimal path in the same grid. We'll see now that a property of this optimal path provides
-
4.1. How to predict gene/protein functions?
RechenmannFrançoisLast week we have seen that annotating a genome means first locating the genes on the DNA sequences that is the genes, the region coding for proteins. But this is indeed the first step,the next very
-
4.10. How efficient is this algorithm?
RechenmannFrançoisWe have seen the principle of an iterative algorithm in two paths for aligning and comparing two sequences of characters, here DNA sequences. And we understoodwhy the iterative version is much more
-
4.4. Aligning sequences is an optimization problem
RechenmannFrançoisWe have seen a nice and a quitesimple solution for measuring the similarity between two sequences. It relied on the so-called hammingdistance that is counting the number of differencesbetween two
-
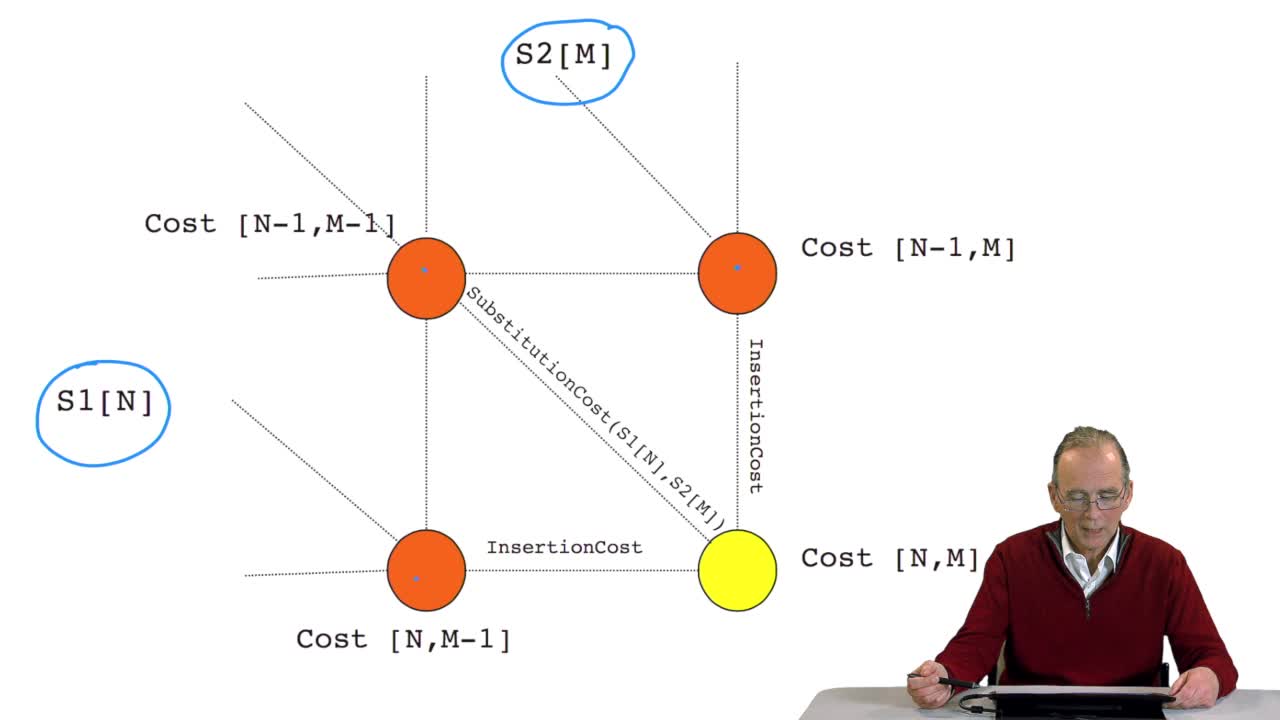
4.7. Alignment costs
RechenmannFrançoisWe have seen how we can compute the cost of the path ending on the last node of our grid if we know the cost of the sub-path ending on the three adjacent nodes. It is time now to see more deeply why
-
4.2. Why gene/protein sequences may be similar?
RechenmannFrançoisBefore measuring the similaritybetween the sequences, it's interesting to answer the question: why gene or protein sequences may be similar? It is indeed veryinteresting because the answer is related
-
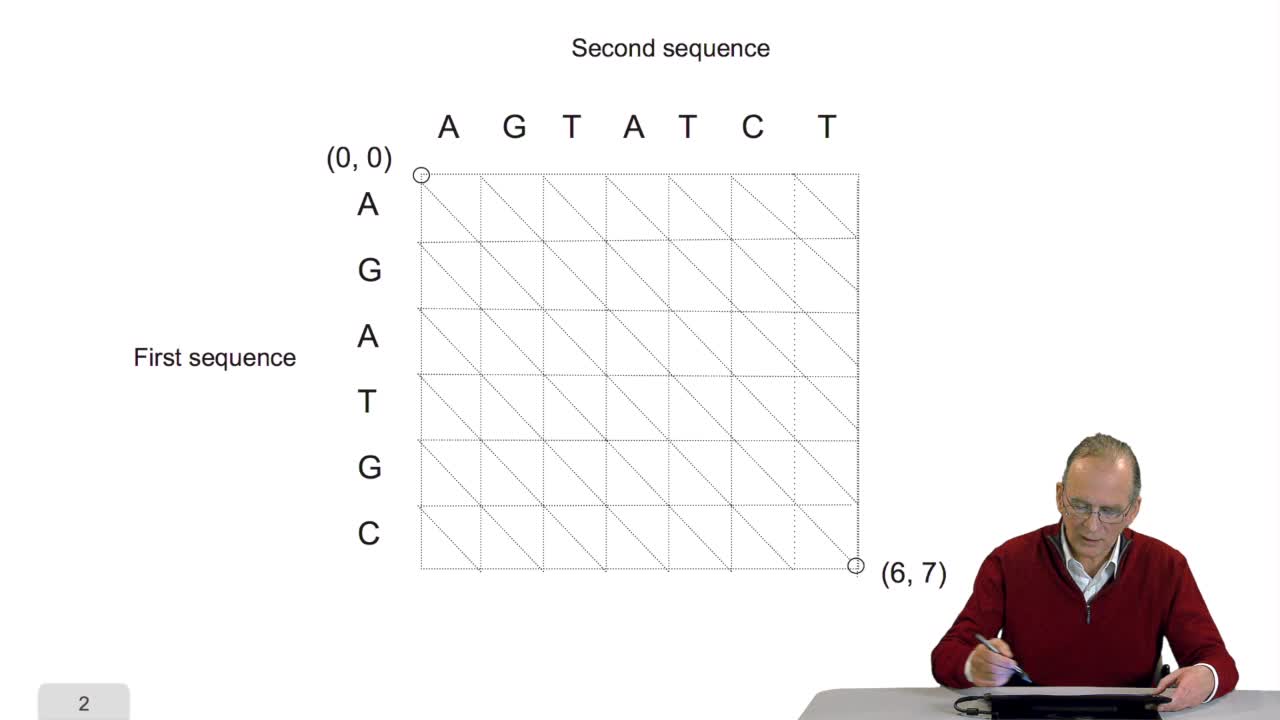
4.5. A sequence alignment as a path
RechenmannFrançoisComparing two sequences and thenmeasuring their similarities is an optimization problem. Why? Because we have seen thatwe have to take into account substitution and deletion. During the alignment, the
-
4.8. A recursive algorithm
RechenmannFrançoisWe have seen how we can computethe optimal cost, the ending node of our grid if we know the optimal cost of the three adjacent nodes. This is this computation scheme we can see here using the notation
-
4.3. Measuring sequence similarity
RechenmannFrançoisSo we understand why gene orprotein sequences may be similar. It's because they evolve togetherwith the species and they evolve in time, there aremodifications in the sequence and that the sequence






Avec les mêmes intervenants et intervenantes
-
1.4. What is an algorithm?
RechenmannFrançoisWe have seen that a genomic textcan be indeed a very long sequence of characters. And to interpret this sequence of characters, we will need to use computers. Using computers means writing program.
-
2.2. Genes: from Mendel to molecular biology
RechenmannFrançoisThe notion of gene emerged withthe works of Gregor Mendel. Mendel studied the inheritance on some traits like the shape of pea plant seeds,through generations. He stated the famous laws of inheritance
-
2.10. How to find genes?
RechenmannFrançoisGetting the sequence of the genome is only the beginning, as I explained, once you have the sequence what you want to do is to locate the gene, to predict the function of the gene and maybe study the
-
3.8. Probabilistic methods
RechenmannFrançoisUp to now, to predict our gene,we only rely on the process of searching certain strings or patterns. In order to further improve our gene predictor, the idea is to use, to rely onprobabilistic methods
-
4.3. Measuring sequence similarity
RechenmannFrançoisSo we understand why gene orprotein sequences may be similar. It's because they evolve togetherwith the species and they evolve in time, there aremodifications in the sequence and that the sequence
-
5.4. The UPGMA algorithm
RechenmannFrançoisWe know how to fill an array with the values of the distances between sequences, pairs of sequences which are available in the file. This array of distances will be the input of our algorithm for
-
1.7. DNA walk
RechenmannFrançoisWe will now design a more graphical algorithm which is called "the DNA walk". We shall see what does it mean "DNA walk". Walk on to DNA. Something like that, yes. But first, just have a look again at
-
2.6. Algorithms + data structures = programs
RechenmannFrançoisBy writing the Lookup GeneticCode Function, we completed our translation algorithm. So we may ask the question about the algorithm, does it terminate? Andthe answer is yes, obviously. Is it pertinent,
-
3.3. Searching for start and stop codons
RechenmannFrançoisWe have written an algorithm for finding genes. But you remember that we arestill to write the two functions for finding the next stop codonand the next start codon. Let's see how we can do that. We
-
4.1. How to predict gene/protein functions?
RechenmannFrançoisLast week we have seen that annotating a genome means first locating the genes on the DNA sequences that is the genes, the region coding for proteins. But this is indeed the first step,the next very
-
4.10. How efficient is this algorithm?
RechenmannFrançoisWe have seen the principle of an iterative algorithm in two paths for aligning and comparing two sequences of characters, here DNA sequences. And we understoodwhy the iterative version is much more
-
1.2. At the heart of the cell: the DNA macromolecule
RechenmannFrançoisDuring the last session, we saw how at the heart of the cell there's DNA in the nucleus, sometimes of cells, or directly in the cytoplasm of the bacteria. The DNA is what we call a macromolecule, that








