
Parmentelat, Thierry (19..-....)
Thierry Parmentelat a mené une carrière hybride entre les mondes académique et industriel. Ses centres d'intérêt couvrent les langages de programmation, les réseaux, et l'algèbre. Actuellement ingénieur de recherche chez Inria, Thierry Parmentelat utilise Python depuis plus de 10 ans pour ses travaux de recherche, ainsi que pour le développement des plateformes expérimentales dont il a la charge.
- Algorithmes
- Bioinformatique
- Génomique
- biologie application informatique
- ADN
- biologie cellulaire et moléculaire
- modélisation
- biologie application informatique
- biologie cellulaire et moléculaire
- modélisation
- biologie application informatique
- ADN
- biologie cellulaire et moléculaire
- modélisation
- biologie application informatique
Vidéos

1.4. Qu’est-ce qu’un algorithme ?
Les génomes peuvent donc être vus comme une longue suite de lettres écrites dans l'alphabet : A, C, G et T. Comment interpréter ces textes ? Ça va être le sujet de la bio-informatique à l'aide d

2.2. Les gènes, de Mendel à la biologie moléculaire
La séquence de caractères est un bon modèle de l'ADN, un des modèles possibles de l'ADN et il est bon parce qu'il est utile. On va voir en particulier que ce modèle simple peut servir de support à de

3.1. Tous les gènes se terminent sur un codon stop
Une fois la séquence d'un génome complet obtenue, débute la phase d'annotation. L'annotation elle-même consiste tout d'abord à rechercher la localisation, c'est-à-dire la position des gènes sur cette
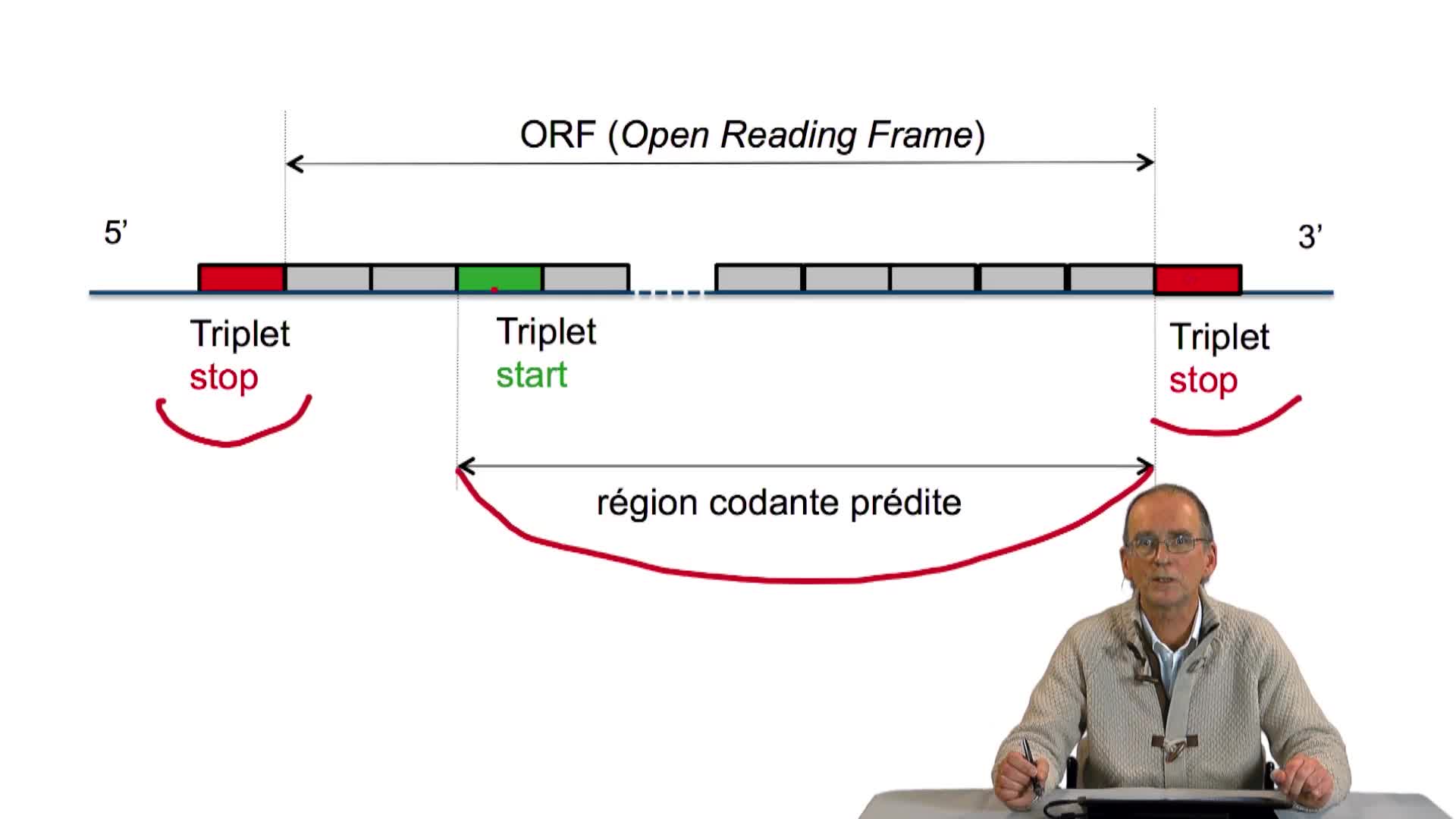
3.2. Un algorithme simple de prédiction de gènes
Sur la base des principes énoncés précédemment, nous allons écrire un premier algorithme de prédiction de gènes sur un texte génomique procaryote. Je rappelle ces principes. L'idée est la suivante :
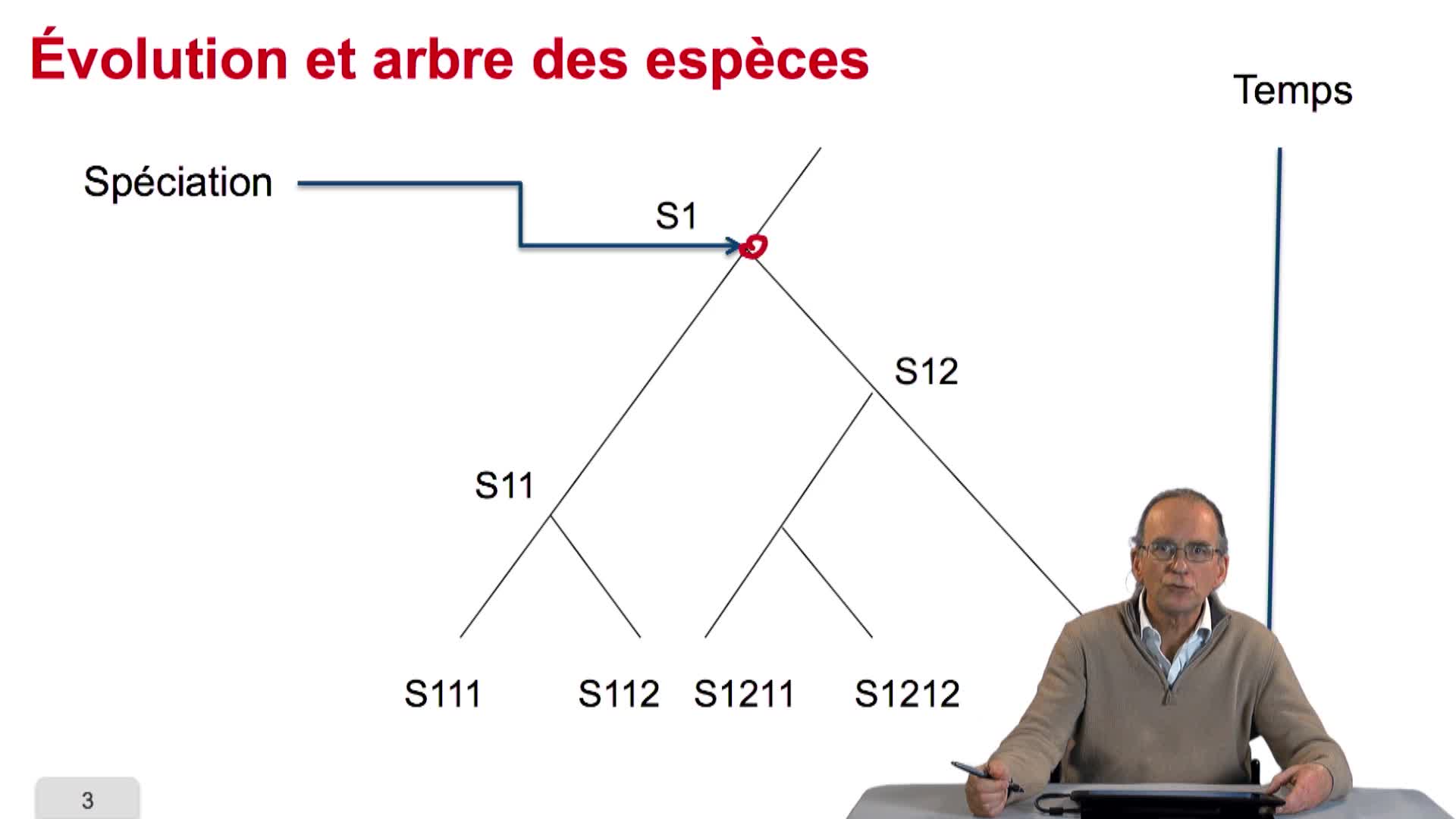
4.2. Évolution et similarité de séquences
Avant de chercher à quantifier ce qu'est la similarité de séquence, on peut se poser la question même de savoir pourquoi des séquences de génome sont similaires entre organismes. La réponse tient dans

4.4. L’alignement de séquences devient un problème d’optimisation
La distance de Hamming nous donne une première possibilité de mesurer la similarité entre 2 séquences. Mais elle ne reflète pas suffisamment la réalité biologique. Qu'est-ce que j'entends par là ? On
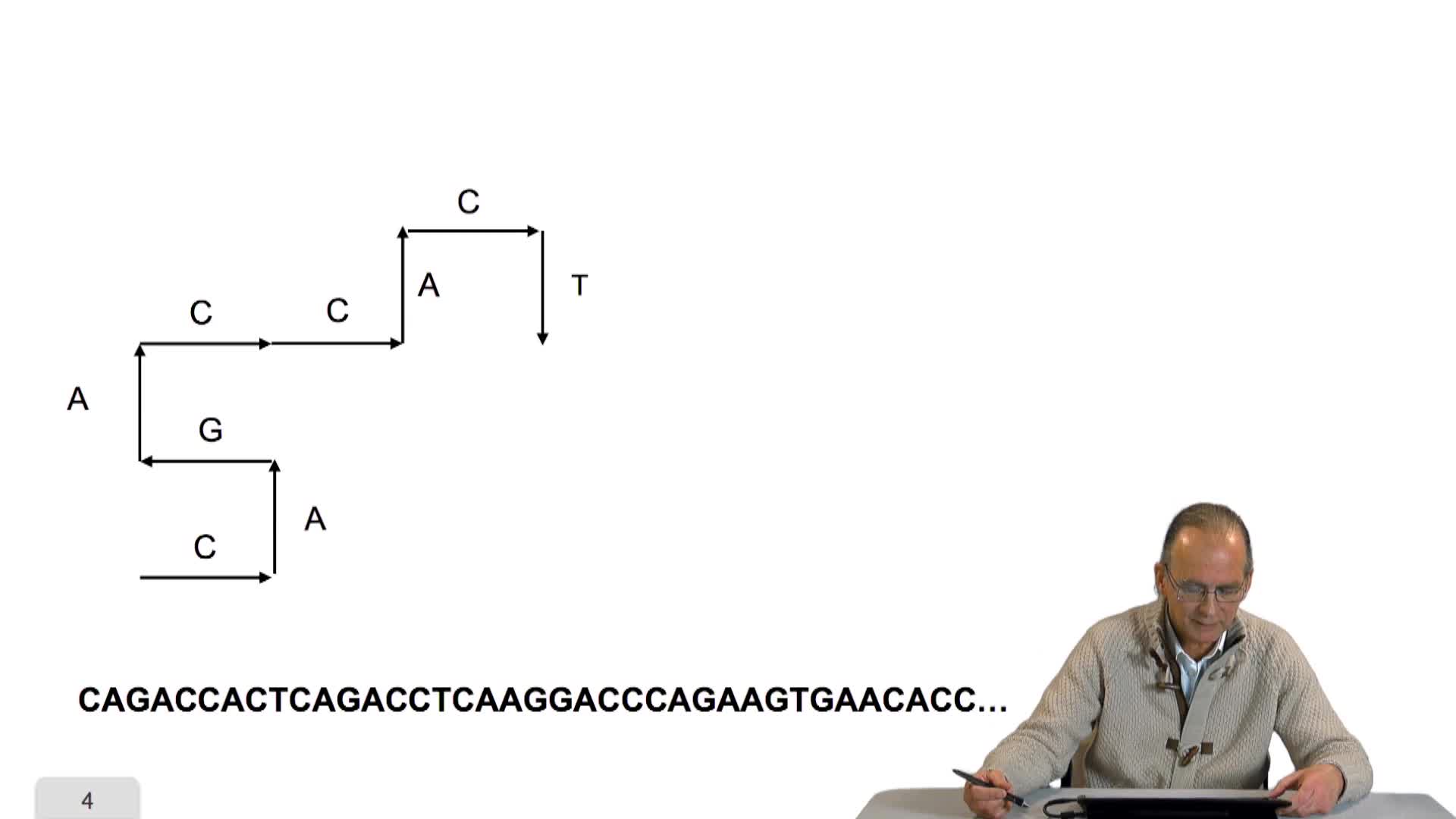
1.8. Changer l’échelle du chemin
Dans la session précédente, je vous ai proposé de m'accompagner dans une balade sur l'ADN. En fait un parcours de la séquence avec un tracé de segments, dont l'orientation dépendait de la lettre
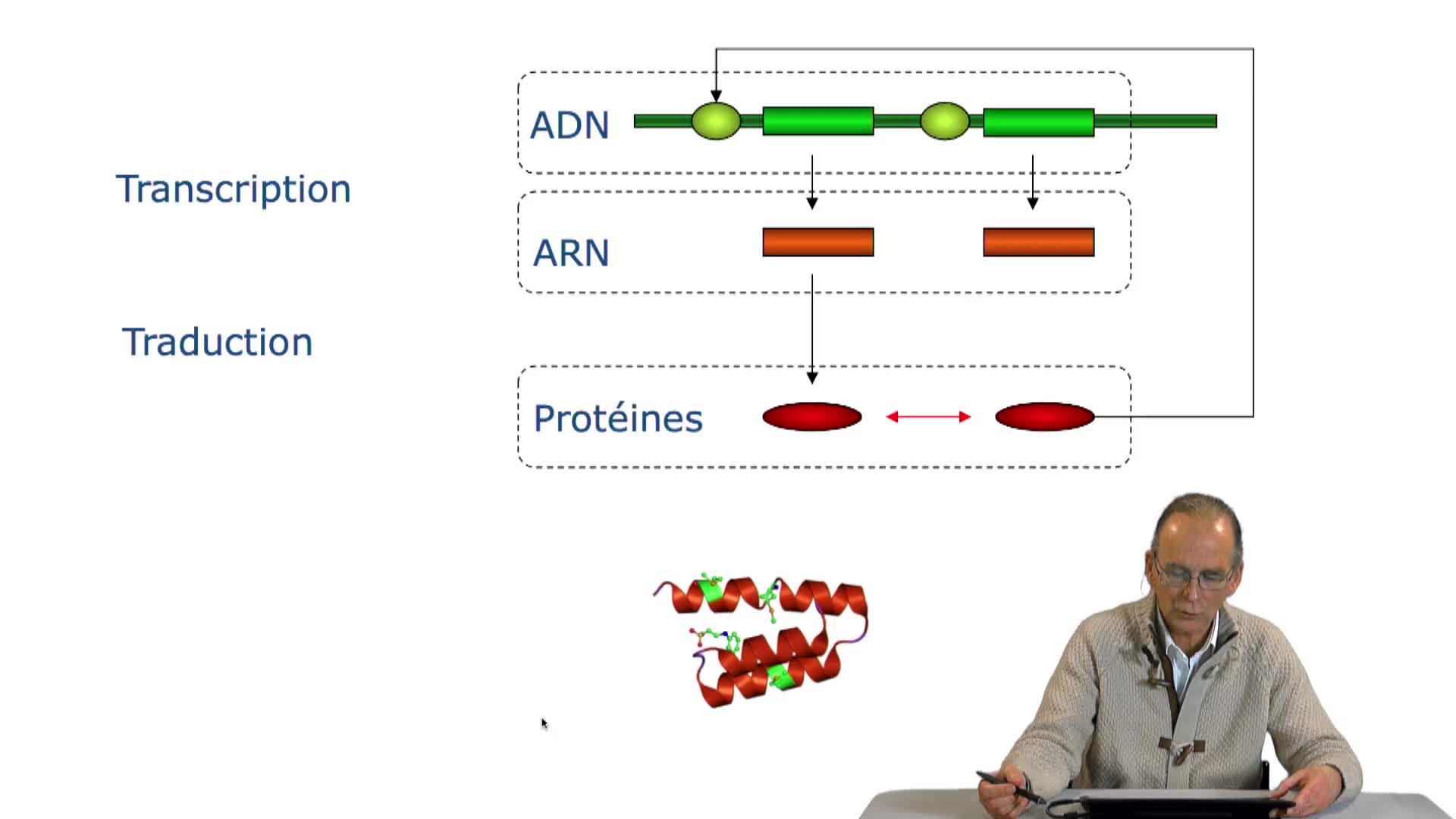
2.3. Le code génétique
Gènes et protéines, mais qu'est-ce qu'une protéine ? Une protéine, c'est également une molécule qui est constituée d'une succession de ce que l'on appelle les acides aminés. C'est donc une chaîne d

3.6. L’algorithme de Boyer-Moore
Vous avez compris que la recherche de motifs, c'est-à-dire de sous-chaînes de caractères dans une chaîne plus importante, était un composant important de beaucoup d'algorithmes de bio-informatique.

4.1. Comment prédire les fonctions des gènes/protéines ?
Après avoir regardé dans les yeux, les semaines précédentes, l'ADN, vu comment cet ADN par séquençage produisait des textes, des séquences génomiques, étudié la relation entre gènes et protéines,

5.4. L’algorithme UPGMA
L'algorithme, que nous allons étudier pour la reconstruction d'arbres phylogénétiques à partir des distances, s'appelle UPGMA. Un nom plutôt compliqué pour une méthode qui est plutôt simple. Et même,

5.6. La diversité des algorithmes informatiques
Nous n'avons vu dans ce cours qu'un exemple extrêmement réduit d'algorithme bio informatique. Il existe en effet une très grande diversité de ces algorithmes bio informatiques qui sont motivés par l