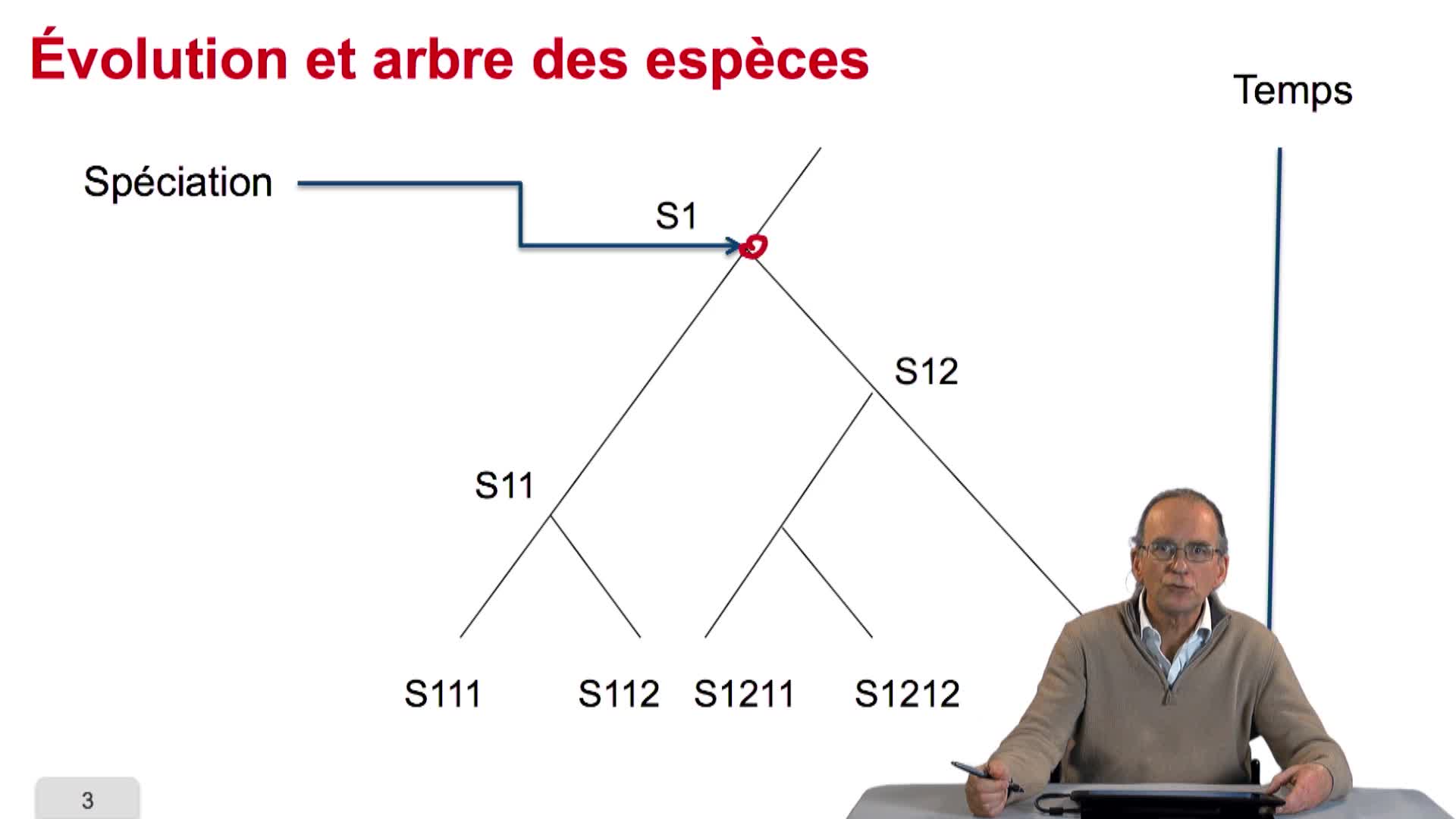
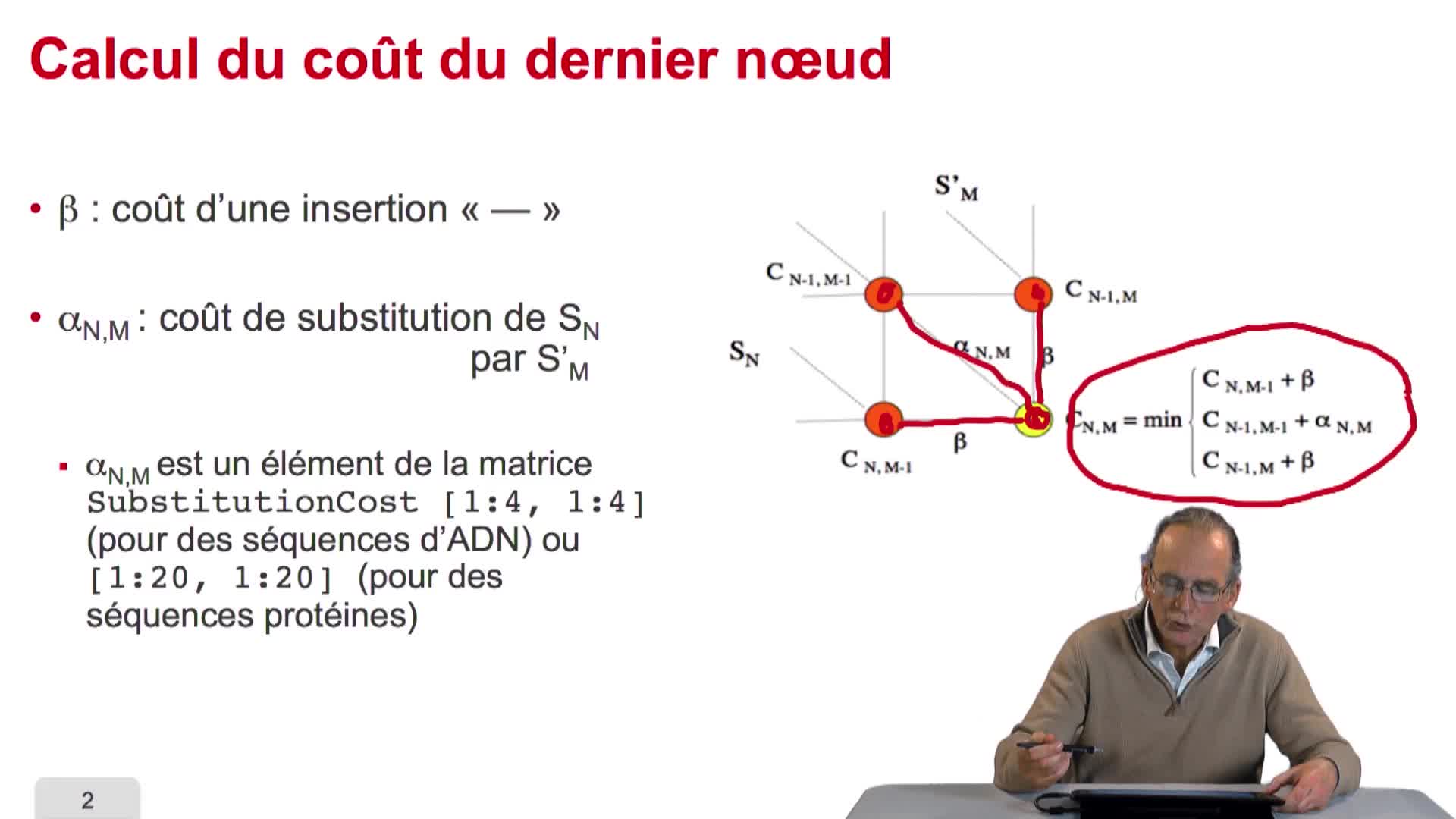
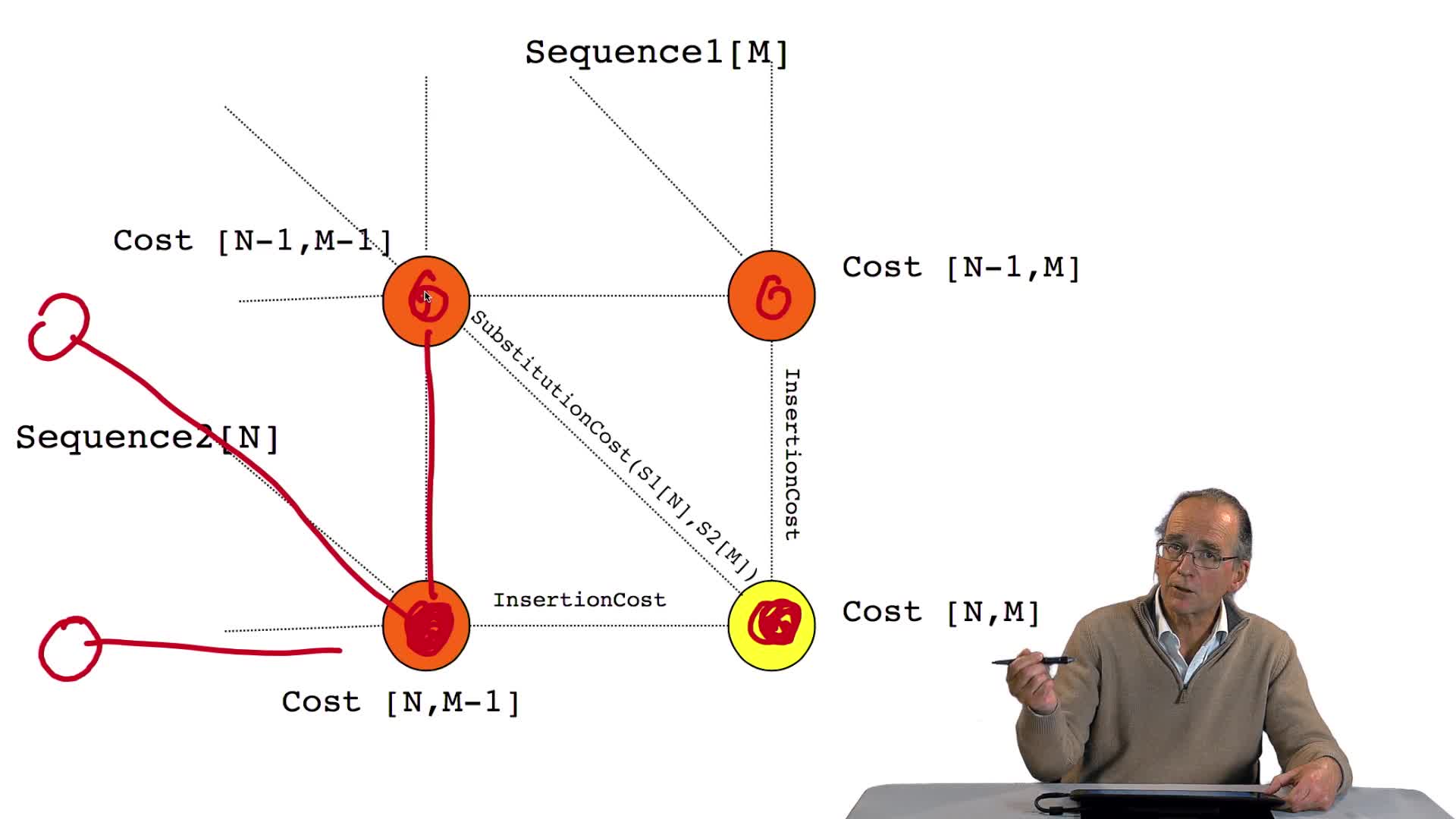
5.3. Remplir un tableau de distances
Rechenmann
François
Parmentelat
Thierry
Pour tenter de construire l'arbre phylogénétique d'un ensemble d'espèces, nous allons utiliser les données et génotypique ou des données génotypiques disponibles sur ces espèces. Plus clairement, nous