Notice
5.4. L’algorithme UPGMA
- document 1 document 2 document 3
- niveau 1 niveau 2 niveau 3
Descriptif
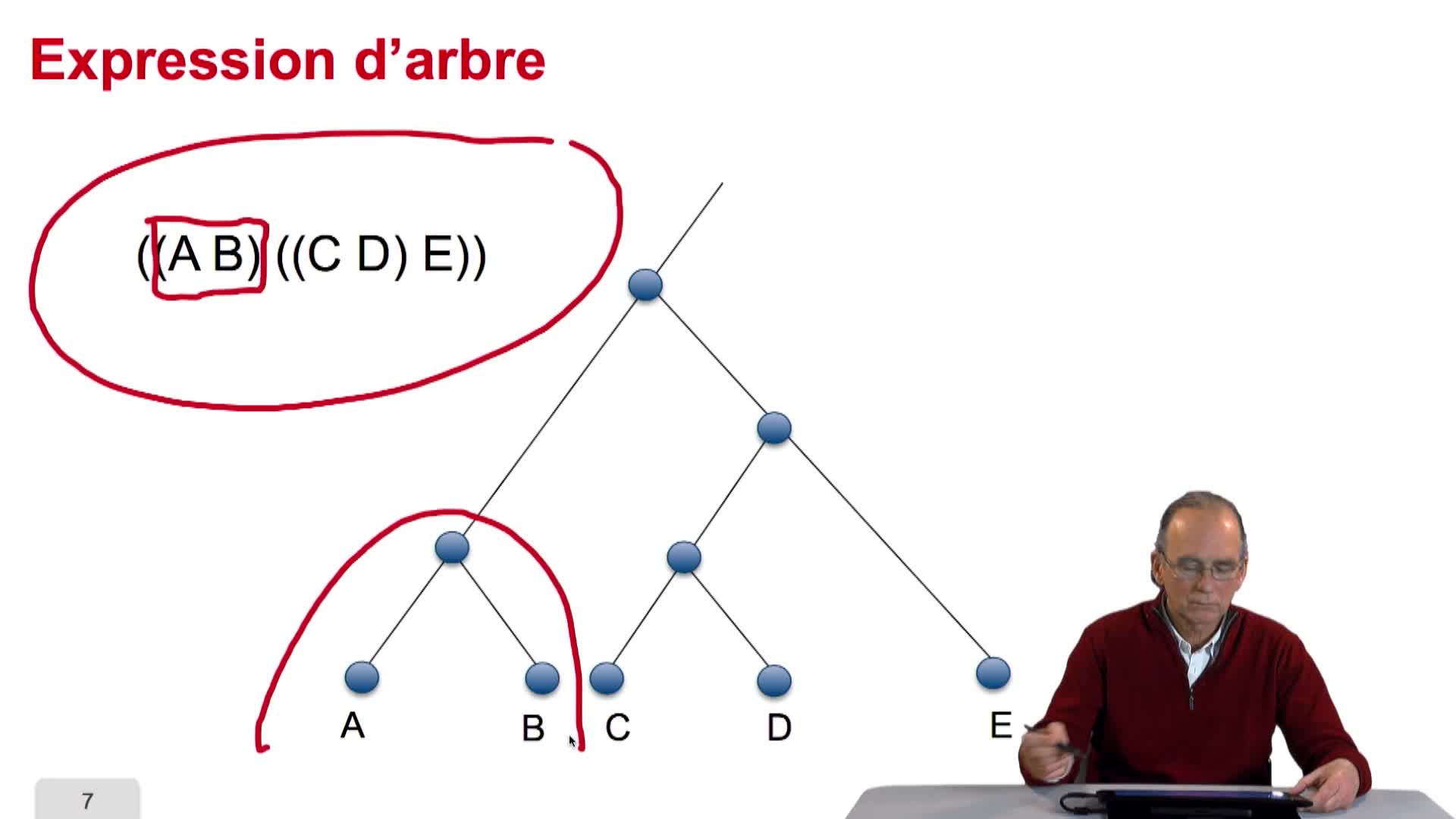
L'algorithme, que nous allons étudier pour la reconstruction d'arbres phylogénétiques à partir des distances, s'appelle UPGMA. Un nom plutôt compliqué pour une méthode qui est plutôt simple. Et même, on le verra trop simple. UPGMA signifie Unweighted Pair Group Method with Arithmetic Mean. Nous allons voir au fur et à mesure, la signification dans l'exécution de l'algorithme de chacun de ces termes. Le point de départ de cet algorithme est donc un tableau de distances, tel que nous avons pu le remplir dans la session précédente. Voilà l'exemple que nous allons traiter. C'est un exemple simple. Nous avons sept espèces différentes et nous avons calculé les distances entre ces espèces à travers le calcul des distances, entre les séquences d'un gène homologue de ces espèces, à toutes ces espèces. Vous vous souvenez que le tableau que nous avons calculé était d'une part symétrique et que d'autre part, les valeurs sur la diagonale étaient sans surprise égales à 0. Ici nous avons choisi de ne conserver et de n'afficher que les valeurs significatives. Donc inutile de montrer les valeurs qui sont les symétriques des autres. Et inutile d'afficher les 0 sur les diagonales. Ce qui explique que notre tableau apparaît incomplet d'une certaine manière. La première étape de l'algorithme consiste à rechercher parmi toutes ces valeurs de distance dans le tableau la plus petite. Ici, c'est 2 et c'est la distance qui sépare l'espèce F de l'espèce C. Raccourci de langage, la distance qui sépare les séquences associées aux espèces F et C. C'est la distance la plus faible. Elle nous pousse donc à grouper ces 2 espèces dans un même sous-graphe en créant un noeud ancêtre ici. Ces 2 espèces sont proches, sont similaires parce qu'elles possèdent un ancêtre commun récent...
ERRATUM
Sur la slide 3 l’orateur parle de 7 espèces différentes, en fait il y en a 6.
Intervention / Responsable scientifique


Dans la même collection
-
5.3. Remplir un tableau de distances
RechenmannFrançoisParmentelatThierryPour tenter de construire l'arbre phylogénétique d'un ensemble d'espèces, nous allons utiliser les données et génotypique ou des données génotypiques disponibles sur ces espèces. Plus clairement, nous
-
5.7. Les applications en microbiologie
RechenmannFrançoisParmentelatThierryUne très grande diversité, on l'a vu, d'algorithmes en bio-informatique, motivé par la résolution de problèmes différents. Ces algorithmes, ces recherches en bio-informatique, s'appuient sur des
-
5.1. L’arbre des espèces
RechenmannFrançoisParmentelatThierryDans cette cinquième et dernière partie de notre cours sur le génome et les algorithmes, qui se veut une introduction à l'analyse informatique de l'information génétique, nous regarderons de plus près
-
5.5. Quand les différences sont trompeuses
RechenmannFrançoisParmentelatThierryIl y a plusieurs raisons pour lesquelles la méthode UPGMA, que nous venons de voir, se révèle simpliste. L'une des raisons par exemple, c'est pourquoi quand on recalcule les distances, quand on a
-
5.2. L’arbre, objet abstrait
RechenmannFrançoisParmentelatThierryVous l'aurez compris un arbre phylogénétique est un arbre abstrait qui n'a qu'un lointain rapport métaphorique avec un véritable arbre. L'arbre des bio-informaticiens et des informaticiens se
-
5.6. La diversité des algorithmes informatiques
RechenmannFrançoisParmentelatThierryNous n'avons vu dans ce cours qu'un exemple extrêmement réduit d'algorithme bio informatique. Il existe en effet une très grande diversité de ces algorithmes bio informatiques qui sont motivés par l





Avec les mêmes intervenants et intervenantes
-
1.6. GC and AT contents of DNA sequence
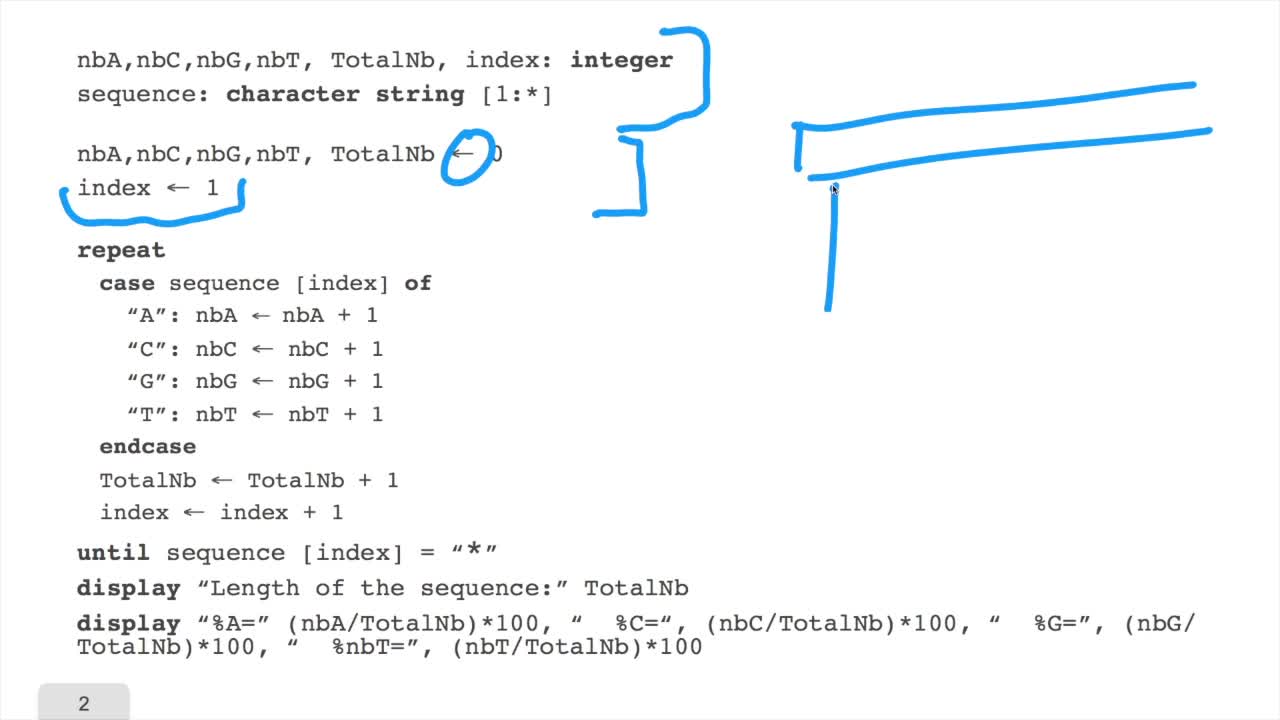
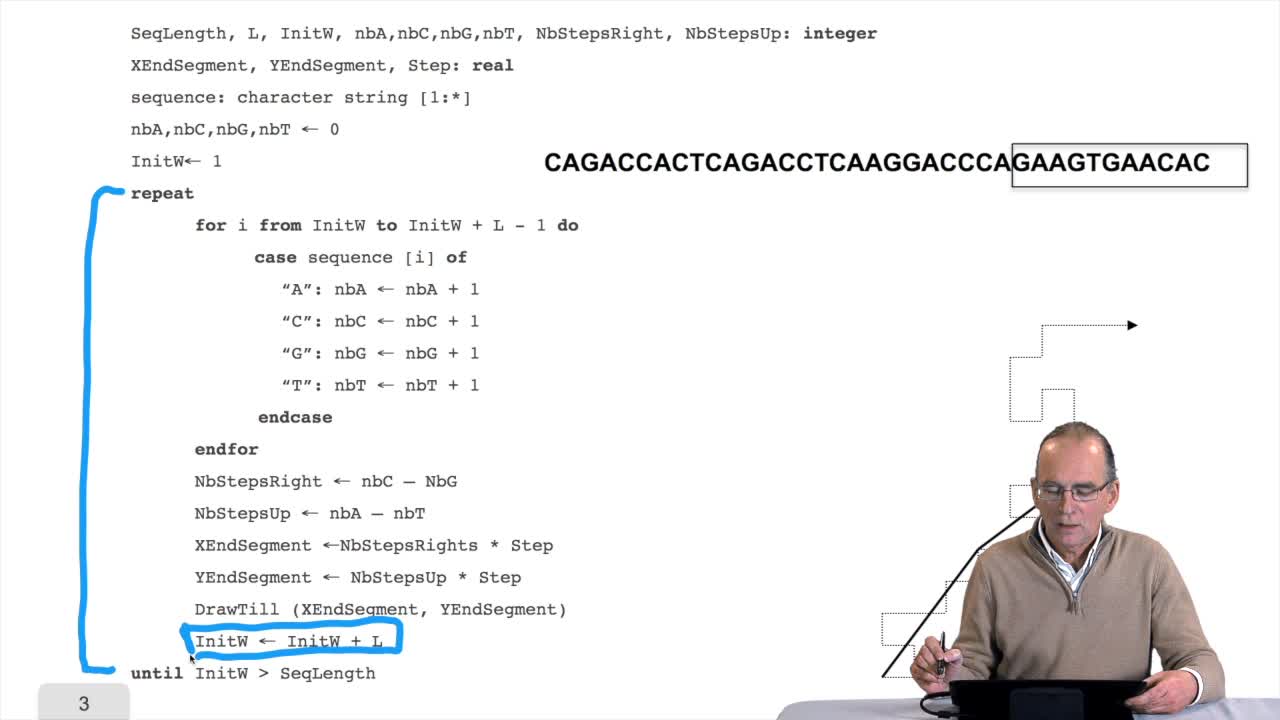
RechenmannFrançoisWe have designed our first algorithmfor counting nucleotides. Remember, what we have writtenin pseudo code is first declaration of variables. We have several integer variables that are variables which
-
2.5. Implementing the genetic code
RechenmannFrançoisRemember we were designing our translation algorithm and since we are a bit lazy, we decided to make the hypothesis that there was the adequate function forimplementing the genetic code. It's now time
-
3.2. A simple algorithm for gene prediction
RechenmannFrançoisBased on the principle we statedin the last session, we will now write in pseudo code a firstalgorithm for locating genes on a bacterial genome. Remember first how this algorithm should work, we first
-
3.10. Gene prediction in eukaryotic genomes
RechenmannFrançoisIf it is possible to have verygood predictions for bacterial genes, it's certainly not the caseyet for eukaryotic genomes. Eukaryotic cells have manydifferences in comparison to prokaryotic cells. You
-
4.8. A recursive algorithm
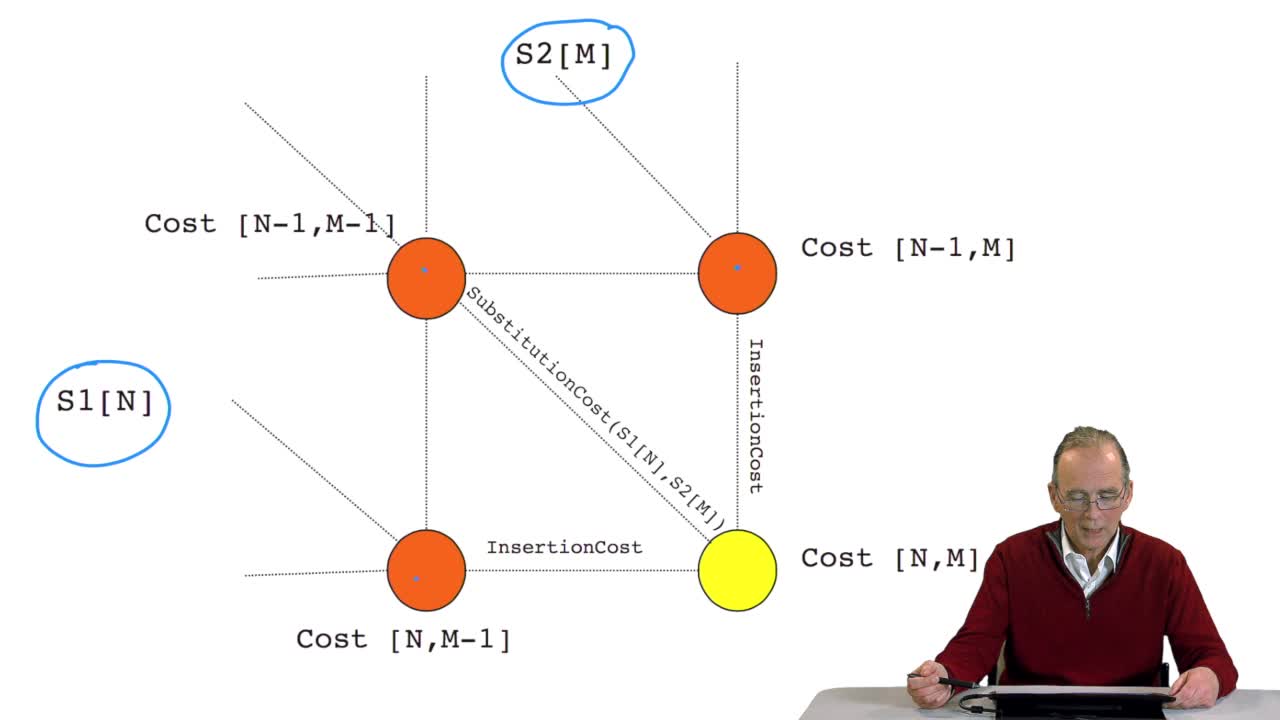
RechenmannFrançoisWe have seen how we can computethe optimal cost, the ending node of our grid if we know the optimal cost of the three adjacent nodes. This is this computation scheme we can see here using the notation
-
5.6. The diversity of bioinformatics algorithms
RechenmannFrançoisIn this course, we have seen a very little set of bioinformatic algorithms. There exist numerous various algorithms in bioinformatics which deal with a large span of classes of problems. For example,
-
1.1. The cell, atom of the living world
RechenmannFrançoisWelcome to this introduction to bioinformatics. We will speak of genomes and algorithms. More specifically, we will see how genetic information can be analysed by algorithms. In these five weeks to
-
1.9. Predicting the origin of DNA replication?
RechenmannFrançoisWe have seen a nice algorithm to draw, let's say, a DNA sequence. We will see that first, we have to correct a little bit this algorithm. And then we will see how such as imple algorithm can provide
-
2.8. DNA sequencing
RechenmannFrançoisDuring the last session, I explained several times how it was important to increase the efficiency of sequences processing algorithm because sequences arevery long and there are large volumes of
-
3.5. Making the predictions more reliable
RechenmannFrançoisWe have got a bacterial gene predictor but the way this predictor works is rather crude and if we want to have more reliable results, we have to inject into this algorithmmore biological knowledge. We
-
4.6. A path is optimal if all its sub-paths are optimal
RechenmannFrançoisA sequence alignment between two sequences is a path in a grid. So that, an optimal sequence alignmentis an optimal path in the same grid. We'll see now that a property of this optimal path provides
-
5.1. The tree of life
RechenmannFrançoisWelcome to this fifth and last week of our course on genomes and algorithms that is the computer analysis of genetic information. During this week, we will firstsee what phylogenetic trees are and how









Sur le même thème
-
La voix, une donnée identifiante à protéger
VincentEmmanuelEmmanuel Vincent, chercheur au Centre Inria de l'Université de Lorraine et au Loria (Laboratoire lorrain de recherche en informatique et ses applications), présente sa recherche sur l'anonymisation de
-
Podcast 1/4 d'heure avec : Emmanuel Vincent, chercheur au Centre Inria de l'Université de Lorraine …
VincentEmmanuelRencontre avec Emmanuel Vincent - chercheur au Centre Inria de l'Université de Lorraine et Loria (Laboratoire lorrain de recherche en informatique et ses applications).
-
Stockage de données numériques sur ADN synthétique : Introduction au domaine
AntoniniMarcDuprazElsaLavenierDominiquePrésentation globale des différentes étapes du stockage de données sur des molécules d'ADN synthétique
-
Stockage de données numériques sur ADN synthétique : Production des données: synthèse, séquençage
LavenierDominiqueBarbryPascalDescription des opérations d'écriture et de lecture des molécules d'ADN : synthèse et séquençage.
-
Stockage de données numériques sur ADN synthétique : Reconstruction des données
LavenierDominiqueTraitement des données après séquençage
-
Stockage de données numériques sur ADN synthétique : Codage Canal
DuprazElsaTechniques de codage pour le stockage de données sur ADN
-
Stockage de données numériques sur ADN synthétique : Codage Source
AntoniniMarcCodage source pour le stockage de données sur ADN synthétique
-
Stockage de données numériques sur ADN synthétique : Théorie de l'information
Kas HannaSergeQuelle quantité d'information peut-on stocker et récupérer de manière fiable dans l'ADN ?
-
The tree of life
AbbySophieLes Rencontres Exobiologiques pour Doctorants (RED) sont une école de formation sur les « bases de l'astrobiologie ». L’édition 2025 s’est tenue du 16 au 21 mars au Parc Ornithologique du Teich.
-
Machines algorithmiques, mythes et réalités
MazenodVincentVincent Mazenod, informaticien, partage le fruit de ses réflexions sur l'évolution des outils numériques, en lien avec les problématiques de souveraineté, de sécurité et de vie privée...
-
Désassemblons le numérique - #Episode11 : Les algorithmes façonnent-ils notre société ?
SchwartzArnaudLima PillaLaércioEstériePierreSalletFrédéricFerbosAudeRoumanosRayyaChraibi KadoudIkramUn an après le tout premier hackathon sur les méthodologies d'enquêtes journalistiques sur les algorithmes, ce nouvel épisode part à la rencontre de différents points de vue sur les algorithmes.
-
Les machines à enseigner. Du livre à l'IA...
BruillardÉricQue peut-on, que doit-on déléguer à des machines ? C'est l'une des questions explorées par Éric Bruillard qui, du livre aux IA génératives, expose l'évolution des machines à enseigner...











