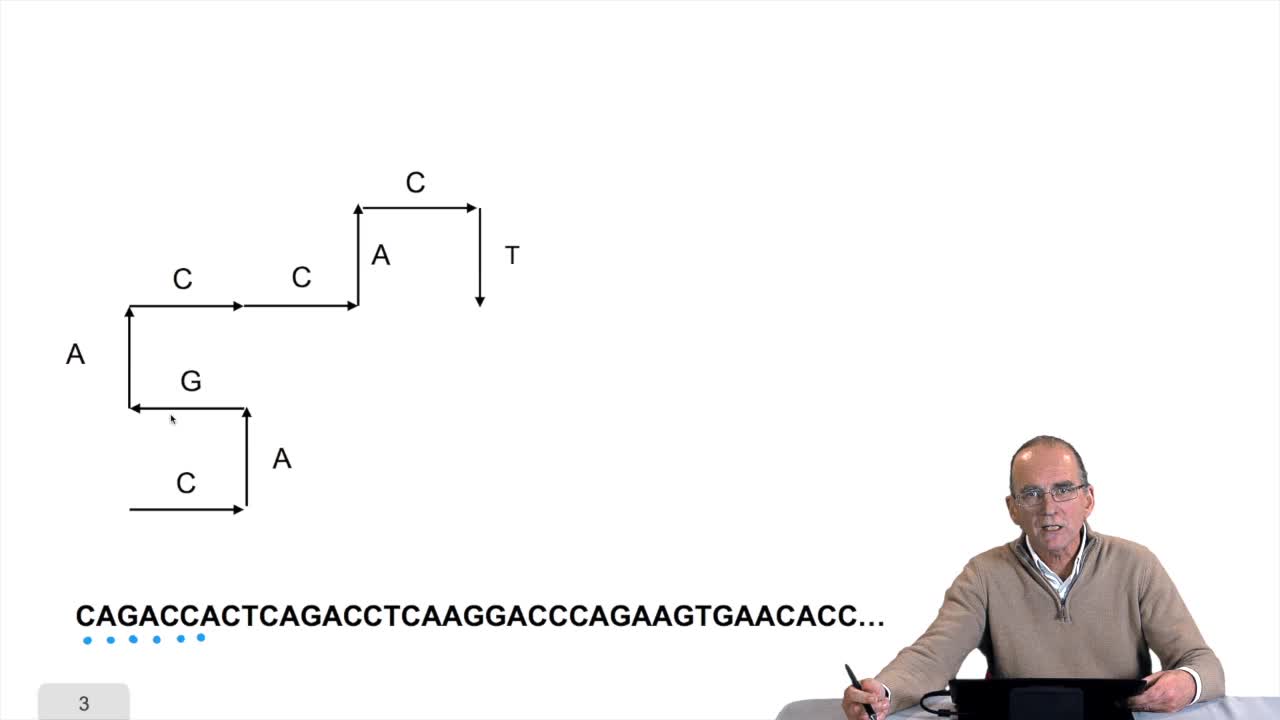Notice
2.9. Whole genome sequencing
- document 1 document 2 document 3
- niveau 1 niveau 2 niveau 3
Descriptif
Sequencing is anexponential technology. The progresses in this technologyallow now to a sequence whole genome, complete genome. What does it mean? Well let'stake two examples: some twenty years ago, to sequence the bacillus subtilis bacteria genome took something like ten years,thirty five laboratories and several millions of euros. It was partly a European project,now some hundreds of dollars and it can be done within a day. The human genome project, famoushuman genome project, more than ten years, three billiondollars, 19-91 dollars OK.
Tomorrow certainly less than 1000 dollars per genome, it means that we can now sequence humangenomes, not one but many many human genomes for the sake ofcomparison, diagnosis and so on. So this is a good measure in theevolution of the sequencing technologies, a capacity ofsequencing entire genomes, not part of them, not one gene here, onegene there, the whole genome, to have access to the entiregenetic information of a living organism being a virus, bacteria,a human being, a plant and so on. Let's see what the ordersof magnitudes are. A virus like the influenza is ten tothe power of four letters nucleotides. The bacteria, typical bacteria Escherichia coli, E. coli in English: ten tothe power of six letters. Yeast, famous yeast, you certainly appreciate yeast, it's for beer for example: ten tothe power of seven.
Intervention / Responsable scientifique

Thème
Documentation
Dans la même collection
-
2.3. The genetic code
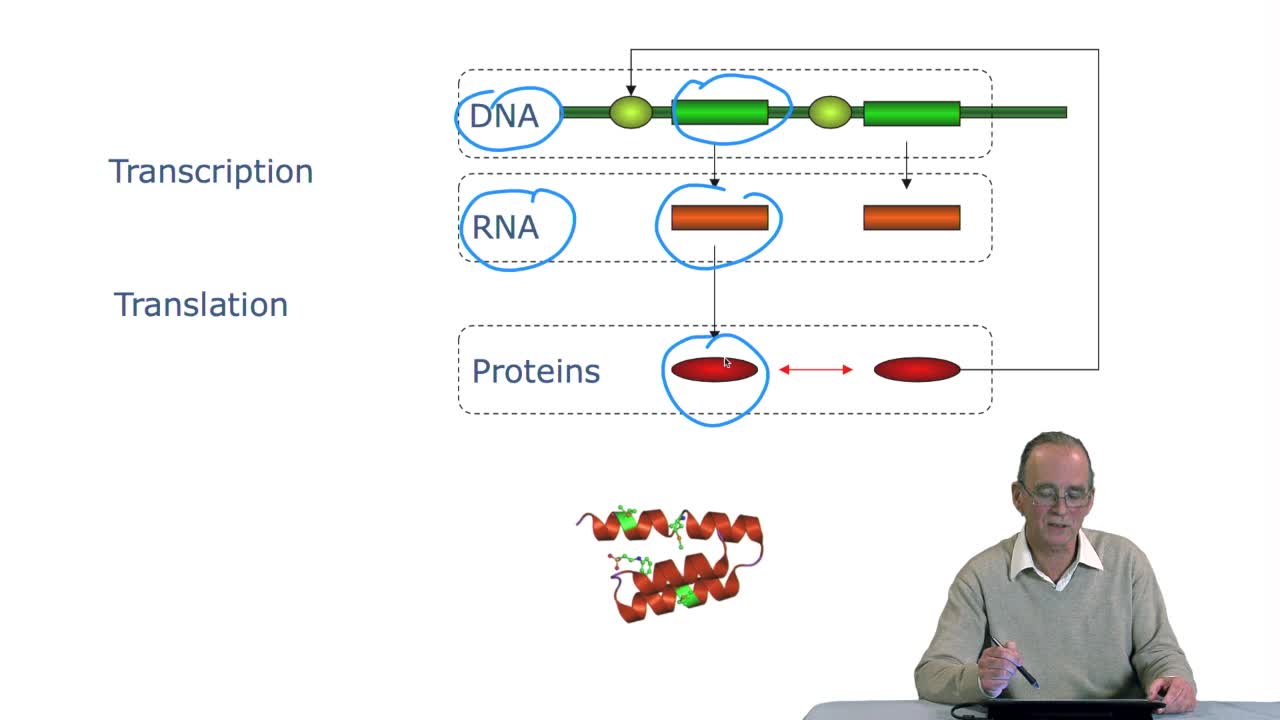
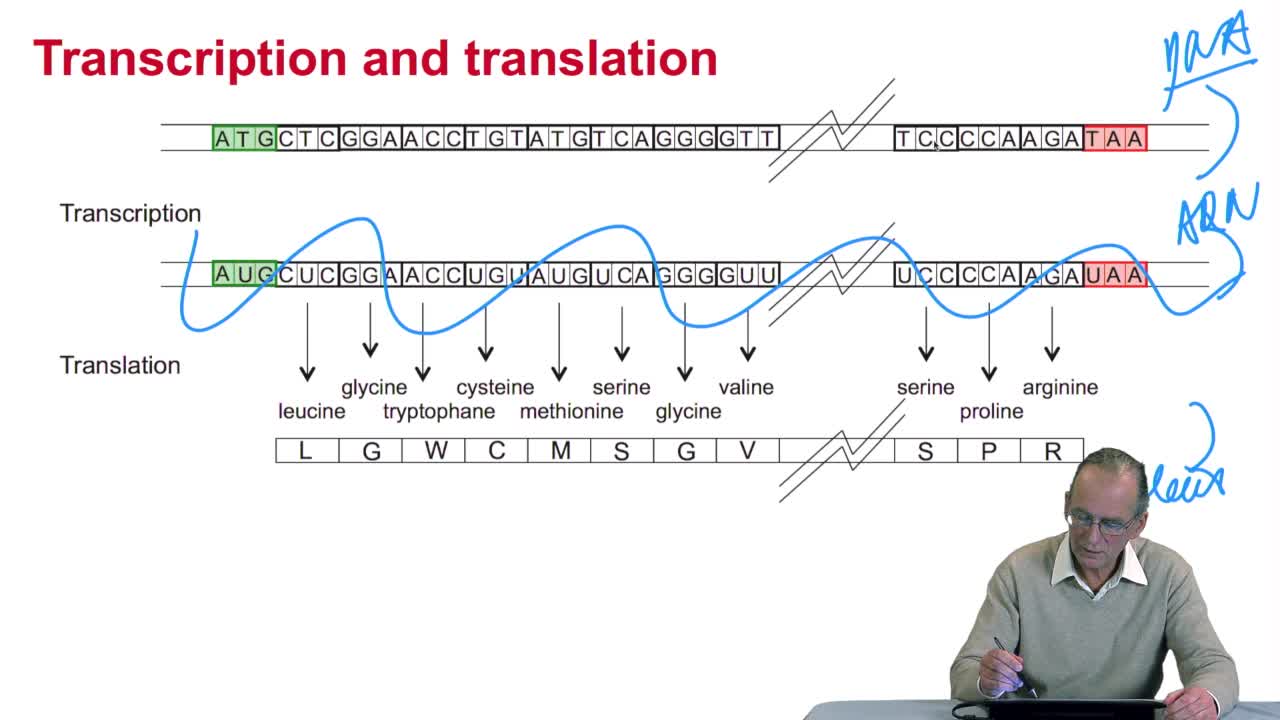
RechenmannFrançoisGenes code for proteins. What is the correspondence betweenthe genes, DNA sequences, and the structure of proteins? The correspondence isthe genetic code. Proteins have indeedsequences of amino acids.
-
2.6. Algorithms + data structures = programs
RechenmannFrançoisBy writing the Lookup GeneticCode Function, we completed our translation algorithm. So we may ask the question about the algorithm, does it terminate? Andthe answer is yes, obviously. Is it pertinent,
-
2.1. The sequence as a model of DNA
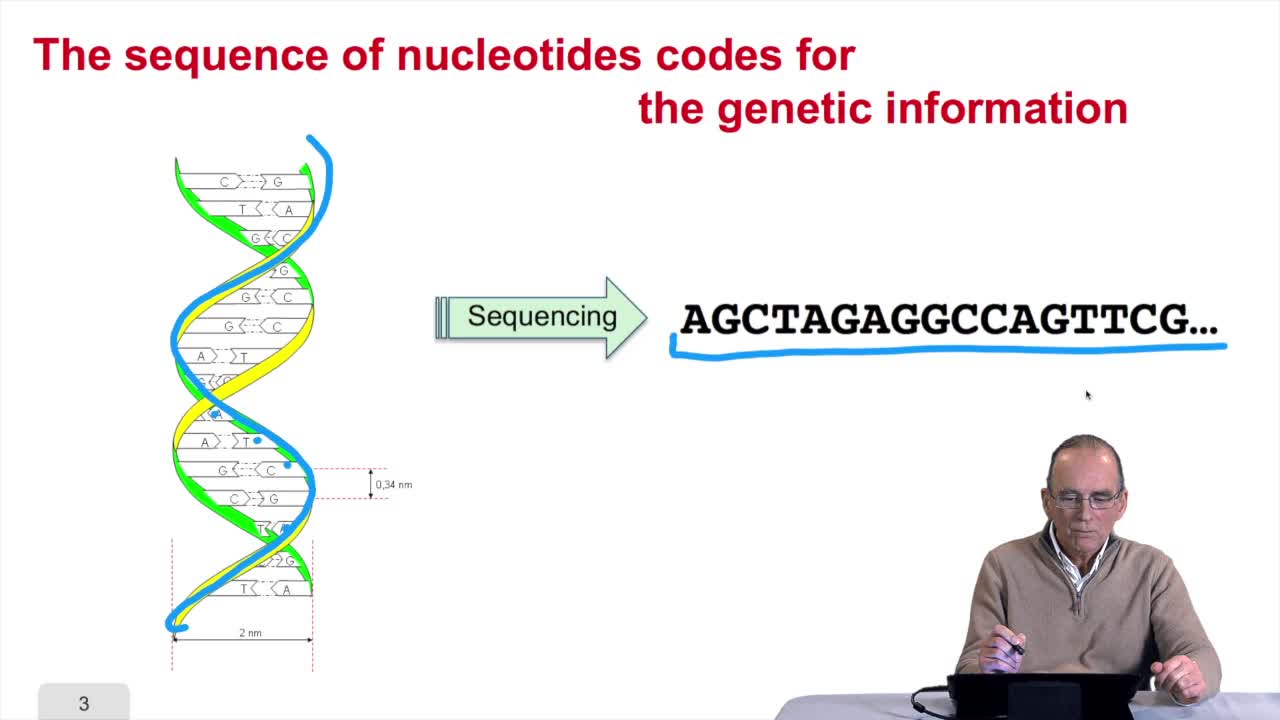
RechenmannFrançoisWelcome back to our course on genomes and algorithms that is a computer analysis ofgenetic information. Last week we introduced the very basic concept in biology that is cell, DNA, genome, genes
-
2.10. How to find genes?
RechenmannFrançoisGetting the sequence of the genome is only the beginning, as I explained, once you have the sequence what you want to do is to locate the gene, to predict the function of the gene and maybe study the
-
2.4. A translation algorithm
RechenmannFrançoisWe have seen that the genetic codeis a correspondence between the DNA or RNA sequences and aminoacid sequences that is proteins. Our aim here is to design atranslation algorithm, we make the
-
2.7. The algorithm design trade-off
RechenmannFrançoisWe saw how to increase the efficiencyof our algorithm through the introduction of a data structure. Now let's see if we can do even better. We had a table of index and weexplain how the use of these
-
2.2. Genes: from Mendel to molecular biology
RechenmannFrançoisThe notion of gene emerged withthe works of Gregor Mendel. Mendel studied the inheritance on some traits like the shape of pea plant seeds,through generations. He stated the famous laws of inheritance
-
2.5. Implementing the genetic code
RechenmannFrançoisRemember we were designing our translation algorithm and since we are a bit lazy, we decided to make the hypothesis that there was the adequate function forimplementing the genetic code. It's now time
-
2.8. DNA sequencing
RechenmannFrançoisDuring the last session, I explained several times how it was important to increase the efficiency of sequences processing algorithm because sequences arevery long and there are large volumes of







Avec les mêmes intervenants et intervenantes
-
1.8. Compressing the DNA walk
RechenmannFrançoisWe have written the algorithm for the circle DNA walk. Just a precision here: the kind of drawing we get has nothing to do with the physical drawing of the DNA molecule. It is a symbolic
-
2.7. The algorithm design trade-off
RechenmannFrançoisWe saw how to increase the efficiencyof our algorithm through the introduction of a data structure. Now let's see if we can do even better. We had a table of index and weexplain how the use of these
-
3.5. Making the predictions more reliable
RechenmannFrançoisWe have got a bacterial gene predictor but the way this predictor works is rather crude and if we want to have more reliable results, we have to inject into this algorithmmore biological knowledge. We
-
4.6. A path is optimal if all its sub-paths are optimal
RechenmannFrançoisA sequence alignment between two sequences is a path in a grid. So that, an optimal sequence alignmentis an optimal path in the same grid. We'll see now that a property of this optimal path provides
-
5.1. The tree of life
RechenmannFrançoisWelcome to this fifth and last week of our course on genomes and algorithms that is the computer analysis of genetic information. During this week, we will firstsee what phylogenetic trees are and how
-
1.3. DNA codes for genetic information
RechenmannFrançoisRemember at the heart of any cell,there is this very long molecule which is called a macromolecule for this reason, which is the DNA molecule. Now we will see that DNA molecules support what is called
-
2.1. The sequence as a model of DNA
RechenmannFrançoisWelcome back to our course on genomes and algorithms that is a computer analysis ofgenetic information. Last week we introduced the very basic concept in biology that is cell, DNA, genome, genes
-
2.10. How to find genes?
RechenmannFrançoisGetting the sequence of the genome is only the beginning, as I explained, once you have the sequence what you want to do is to locate the gene, to predict the function of the gene and maybe study the
-
3.8. Probabilistic methods
RechenmannFrançoisUp to now, to predict our gene,we only rely on the process of searching certain strings or patterns. In order to further improve our gene predictor, the idea is to use, to rely onprobabilistic methods
-
4.3. Measuring sequence similarity
RechenmannFrançoisSo we understand why gene orprotein sequences may be similar. It's because they evolve togetherwith the species and they evolve in time, there aremodifications in the sequence and that the sequence
-
5.3. Building an array of distances
RechenmannFrançoisSo using the sequences of homologous gene between several species, our aim is to reconstruct phylogenetic tree of the corresponding species. For this, we have to comparesequences and compute distances
-
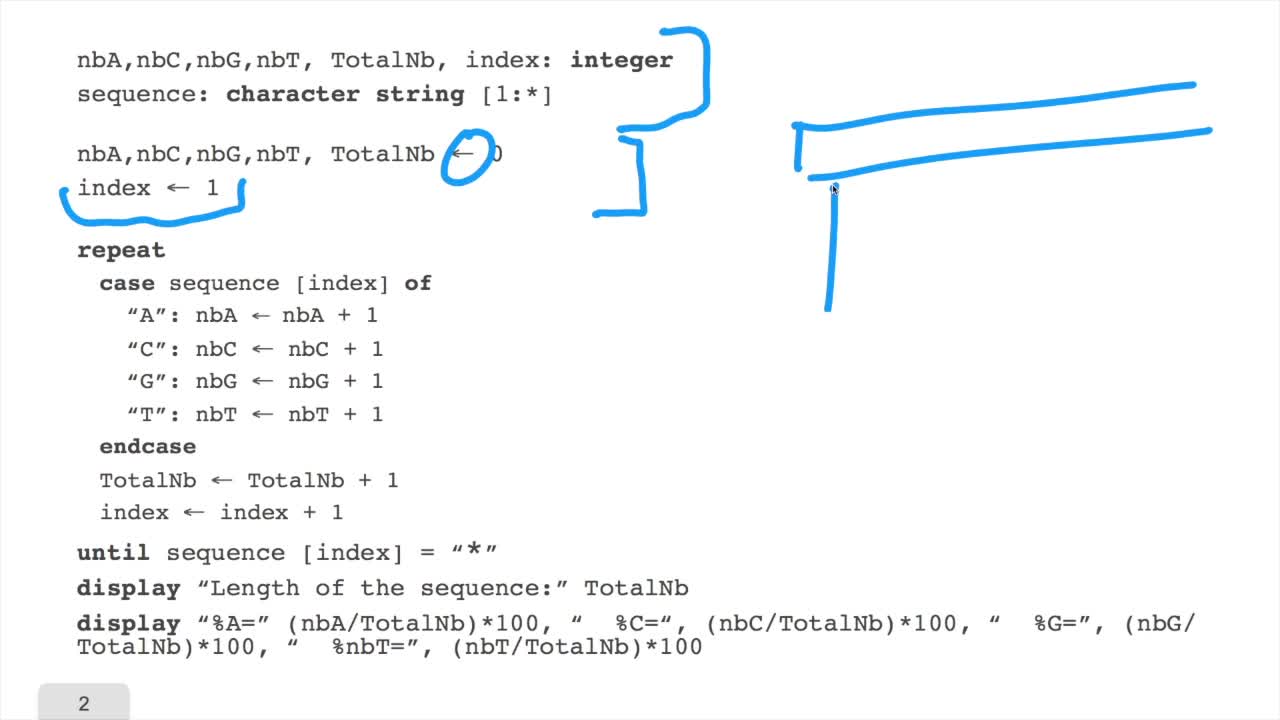
1.6. GC and AT contents of DNA sequence
RechenmannFrançoisWe have designed our first algorithmfor counting nucleotides. Remember, what we have writtenin pseudo code is first declaration of variables. We have several integer variables that are variables which