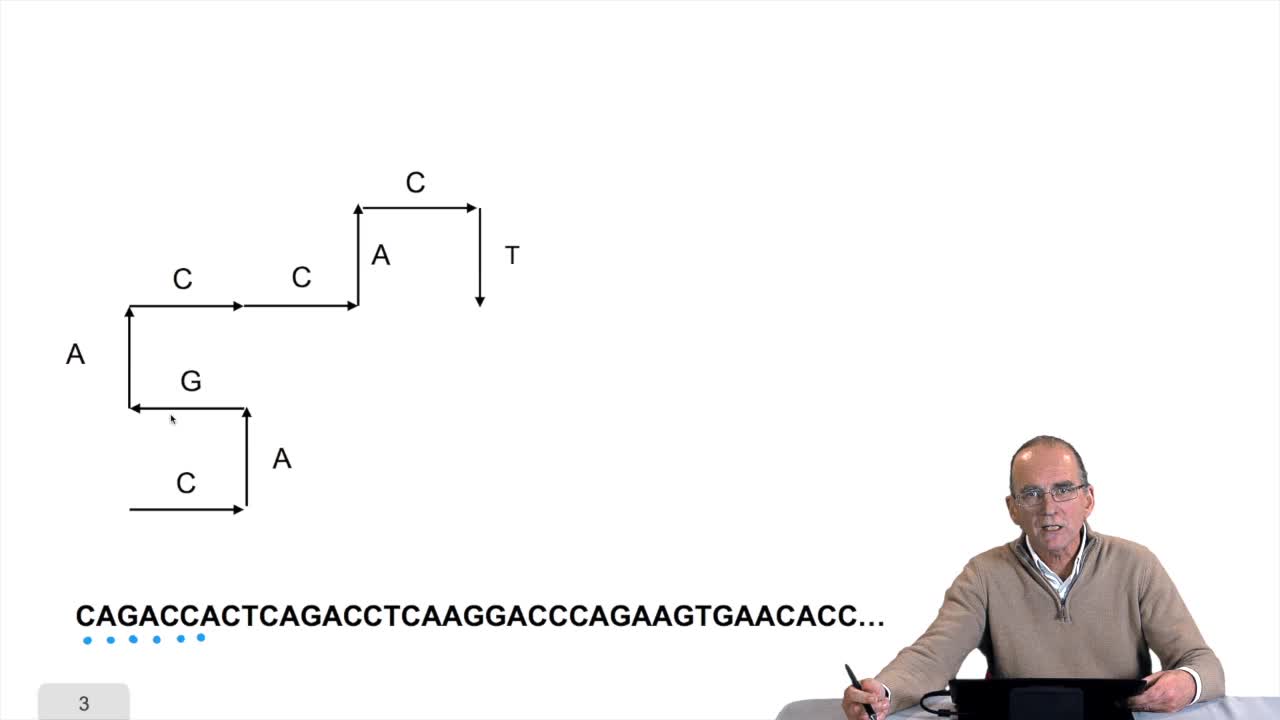Notice
3.9. Benchmarking the prediction methods
- document 1 document 2 document 3
- niveau 1 niveau 2 niveau 3
Descriptif
It is necessary to underline that gene predictors produce predictions. Predictions mean that you have no guarantees that the coding sequences, the coding regions,the genes you get when applying your algorithm, are true genes, thatis genes which have a biological existence. Only experimental analysiscan confirm or infirm your predictions. Nevertheless it is interesting and also important to be able to evaluate your algorithm, thisis the role of benchmarking. Benchmarking means measuring the capacity of your algorithm to produce good predictions. How can we make thiskind of measurement? We need a reference, an idealreference would be a genome which is well annotated and for whichall of the annotations have been confirmed through experimental results. Unfortunately, there are very few genomes for which we have this experimental confirmation.Even, for example, the E. coli genome, which is a well-knownorganism and well annotated genome, is not the ideal reference. However, it is also interesting to compare prediction algorithm and method between them, to do acompetition, to apply several predictors on the same genomes and to compare the results of this algorithm.
Intervention / Responsable scientifique

Thème
Documentation
Dans la même collection
-
3.6. Boyer-Moore algorithm
RechenmannFrançoisWe have seen how we can make gene predictions more reliable through searching for all the patterns,all the occurrences of patterns. We have seen, for example, howif we locate the RBS, Ribosome
-
3.1. All genes end on a stop codon
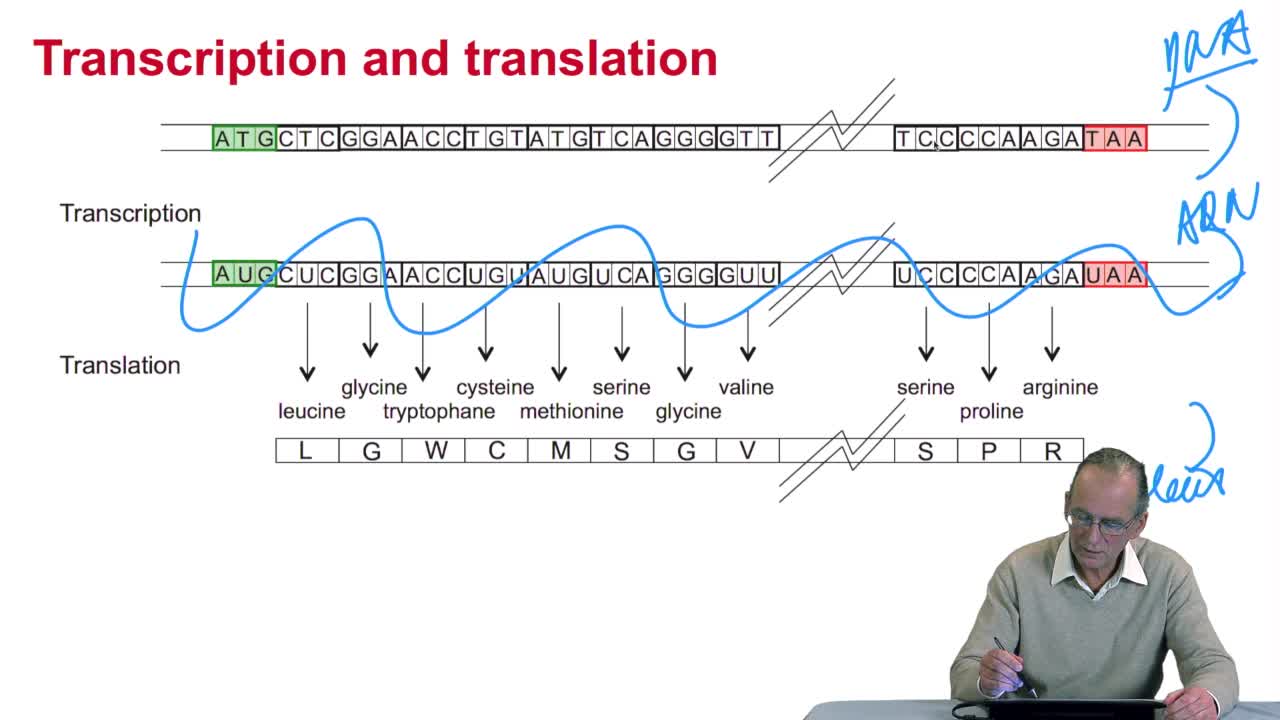
RechenmannFrançoisLast week we studied genes and proteins and so how genes, portions of DNA, are translated into proteins. We also saw the very fast evolutionof the sequencing technology which allows for producing
-
3.10. Gene prediction in eukaryotic genomes
RechenmannFrançoisIf it is possible to have verygood predictions for bacterial genes, it's certainly not the caseyet for eukaryotic genomes. Eukaryotic cells have manydifferences in comparison to prokaryotic cells. You
-
3.4. Predicting all the genes in a sequence
RechenmannFrançoisWe have written an algorithm whichis able to locate potential genes on a sequence but only on one phase because we are looking triplets after triplets. Now remember that the genes maybe located on
-
3.7. Index and suffix trees
RechenmannFrançoisWe have seen with the Boyer-Moore algorithm how we can increase the efficiency of spin searching through the pre-processing of the pattern to be searched. Now we will see that an alternative way of
-
3.2. A simple algorithm for gene prediction
RechenmannFrançoisBased on the principle we statedin the last session, we will now write in pseudo code a firstalgorithm for locating genes on a bacterial genome. Remember first how this algorithm should work, we first
-
3.5. Making the predictions more reliable
RechenmannFrançoisWe have got a bacterial gene predictor but the way this predictor works is rather crude and if we want to have more reliable results, we have to inject into this algorithmmore biological knowledge. We
-
3.8. Probabilistic methods
RechenmannFrançoisUp to now, to predict our gene,we only rely on the process of searching certain strings or patterns. In order to further improve our gene predictor, the idea is to use, to rely onprobabilistic methods
-
3.3. Searching for start and stop codons
RechenmannFrançoisWe have written an algorithm for finding genes. But you remember that we arestill to write the two functions for finding the next stop codonand the next start codon. Let's see how we can do that. We









Avec les mêmes intervenants et intervenantes
-
1.5. Counting nucleotides
RechenmannFrançoisIn this session, don't panic. We will design our first algorithm. This algorithm is forcounting nucleotides. The idea here is that as an input,you have a sequence of nucleotides, of bases, of letters,
-
2.4. A translation algorithm
RechenmannFrançoisWe have seen that the genetic codeis a correspondence between the DNA or RNA sequences and aminoacid sequences that is proteins. Our aim here is to design atranslation algorithm, we make the
-
3.1. All genes end on a stop codon
RechenmannFrançoisLast week we studied genes and proteins and so how genes, portions of DNA, are translated into proteins. We also saw the very fast evolutionof the sequencing technology which allows for producing
-
3.10. Gene prediction in eukaryotic genomes
RechenmannFrançoisIf it is possible to have verygood predictions for bacterial genes, it's certainly not the caseyet for eukaryotic genomes. Eukaryotic cells have manydifferences in comparison to prokaryotic cells. You
-
4.8. A recursive algorithm
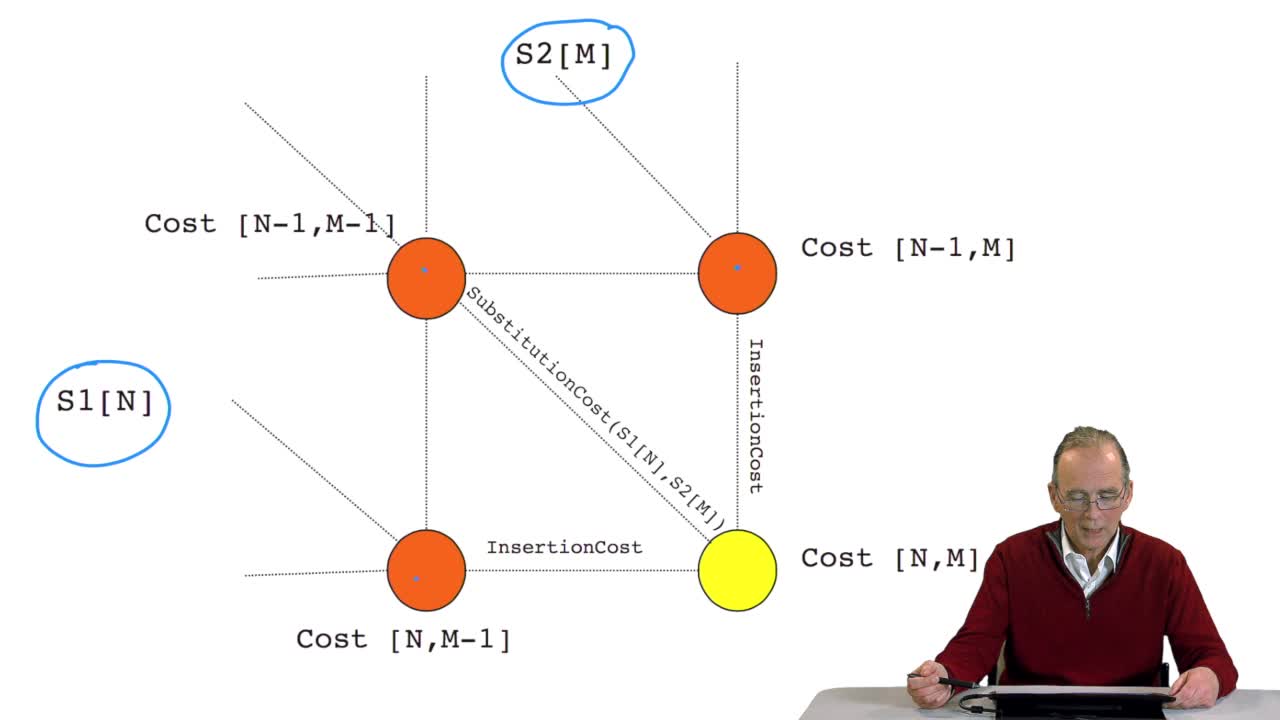
RechenmannFrançoisWe have seen how we can computethe optimal cost, the ending node of our grid if we know the optimal cost of the three adjacent nodes. This is this computation scheme we can see here using the notation
-
5.6. The diversity of bioinformatics algorithms
RechenmannFrançoisIn this course, we have seen a very little set of bioinformatic algorithms. There exist numerous various algorithms in bioinformatics which deal with a large span of classes of problems. For example,
-
1.8. Compressing the DNA walk
RechenmannFrançoisWe have written the algorithm for the circle DNA walk. Just a precision here: the kind of drawing we get has nothing to do with the physical drawing of the DNA molecule. It is a symbolic
-
2.7. The algorithm design trade-off
RechenmannFrançoisWe saw how to increase the efficiencyof our algorithm through the introduction of a data structure. Now let's see if we can do even better. We had a table of index and weexplain how the use of these
-
3.4. Predicting all the genes in a sequence
RechenmannFrançoisWe have written an algorithm whichis able to locate potential genes on a sequence but only on one phase because we are looking triplets after triplets. Now remember that the genes maybe located on
-
4.6. A path is optimal if all its sub-paths are optimal
RechenmannFrançoisA sequence alignment between two sequences is a path in a grid. So that, an optimal sequence alignmentis an optimal path in the same grid. We'll see now that a property of this optimal path provides
-
5.1. The tree of life
RechenmannFrançoisWelcome to this fifth and last week of our course on genomes and algorithms that is the computer analysis of genetic information. During this week, we will firstsee what phylogenetic trees are and how
-
1.3. DNA codes for genetic information
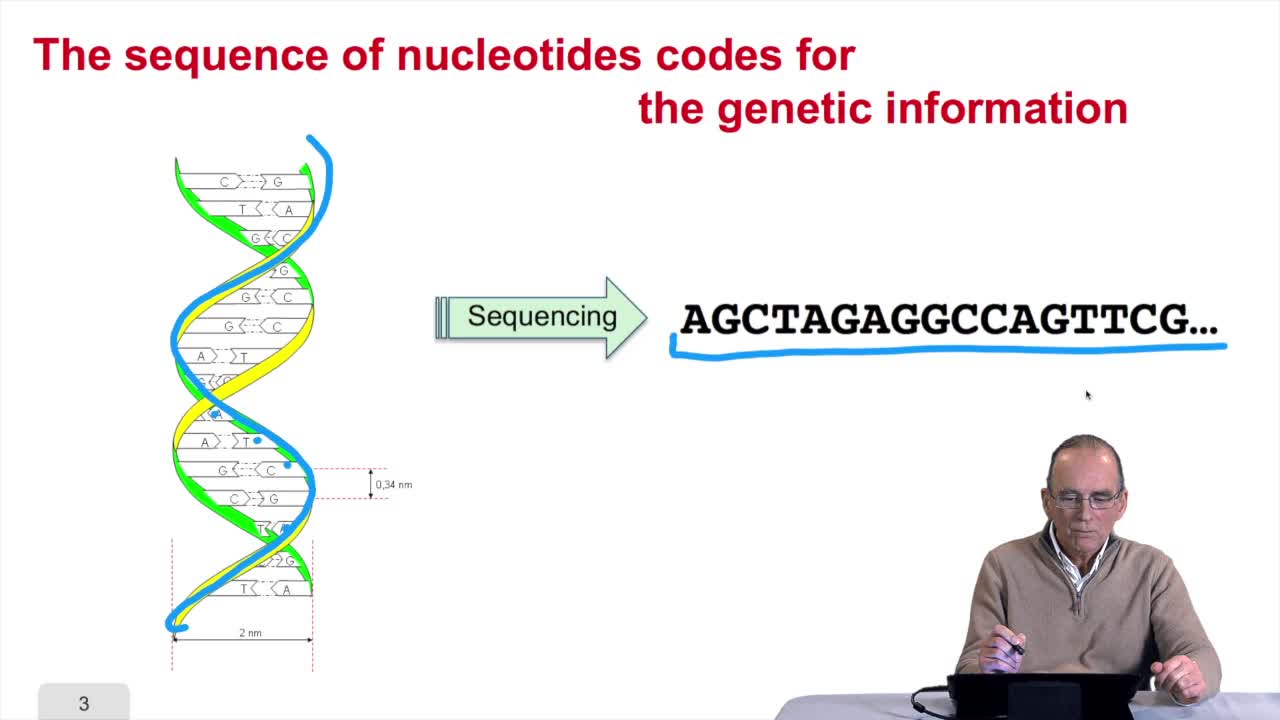
RechenmannFrançoisRemember at the heart of any cell,there is this very long molecule which is called a macromolecule for this reason, which is the DNA molecule. Now we will see that DNA molecules support what is called