Notice
5.1. The tree of life
- document 1 document 2 document 3
- niveau 1 niveau 2 niveau 3
Descriptif
Welcome to this fifth and last week of our course on genomes and algorithms that is the computer analysis of genetic information. During this week, we will firstsee what phylogenetic trees are and how we can reconstruct these trees from the available data. Then to conclude this week and this course, we will present an overview, a larger overview of bioinformatic algorithms and we will conclude on the application of bioinformatics at least in the microbial world. So first the tree of life, we have already seen that due to the ideas of Darwin, we know that species evolve and the evolution of these species canbe seen as a tree. Each note of the tree is aspecies, there are some specific events which are called speciation when one species evolves into two different species and so on. Over time new species appeared and we have at the leaves of the tree the species which are known presently. The problem here is: is it possible to reconstruct the phylogenetic tree of a set of species knowing that we have at our disposal only the data on what we see now? We have to reconstruct a story using the available information and data. The answer is Yes, of course, we have no mean to be sure that the tree we will reconstruct isthe real one but we can make assumptions, hypothesis and usealgorithms to build phylogenetic trees from available information. Two classes of information can beused, the so-called phenotypic information, that is to say, the phenotype is what we see of a living system. The aspect of it, its characteristic, anatomy and so on.
Intervention / Responsable scientifique

Thème
Documentation
Dans la même collection
-
5.3. Building an array of distances
RechenmannFrançoisSo using the sequences of homologous gene between several species, our aim is to reconstruct phylogenetic tree of the corresponding species. For this, we have to comparesequences and compute distances
-
5.6. The diversity of bioinformatics algorithms
RechenmannFrançoisIn this course, we have seen a very little set of bioinformatic algorithms. There exist numerous various algorithms in bioinformatics which deal with a large span of classes of problems. For example,
-
5.4. The UPGMA algorithm
RechenmannFrançoisWe know how to fill an array with the values of the distances between sequences, pairs of sequences which are available in the file. This array of distances will be the input of our algorithm for
-
5.7. The application domains in microbiology
RechenmannFrançoisBioinformatics relies on many domains of mathematics and computer science. Of course, algorithms themselves on character strings are important in bioinformatics, we have seen them. Algorithms and
-
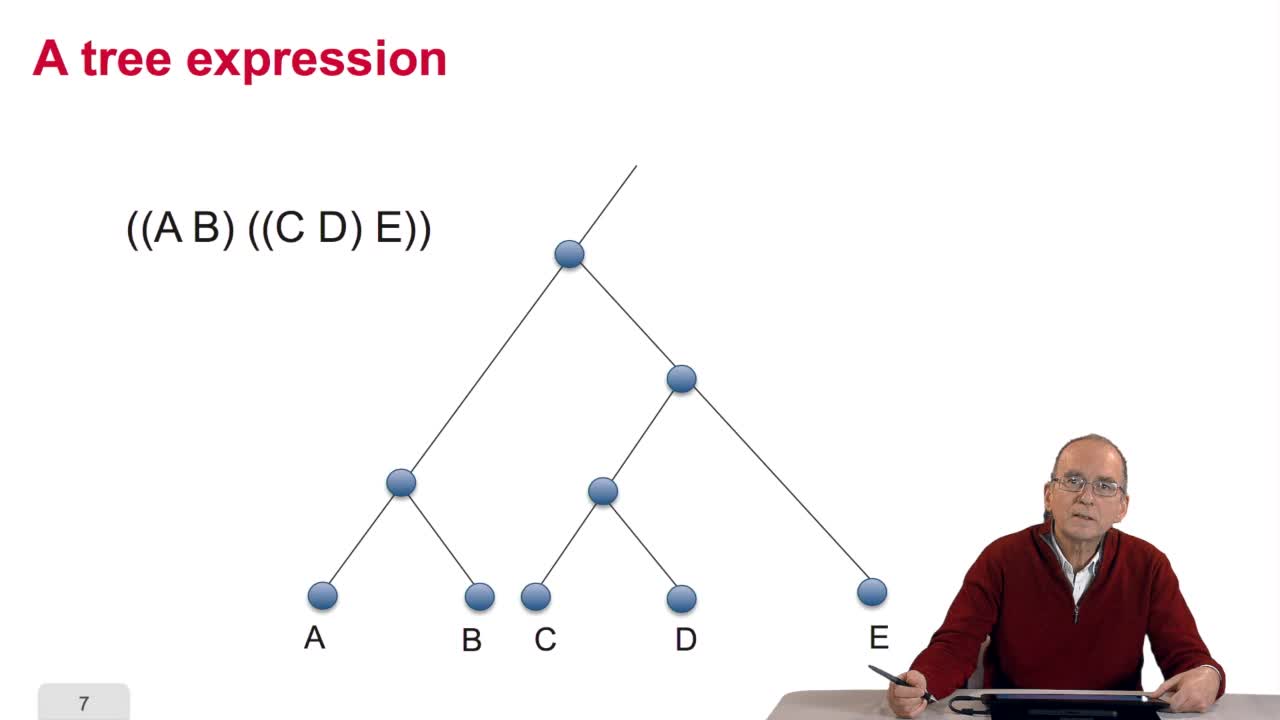
5.2. The tree, an abstract object
RechenmannFrançoisWhen we speak of trees, of species,of phylogenetic trees, of course, it's a metaphoric view of a real tree. Our trees are abstract objects. Here is a tree and the different components of this tree.
-
5.5. Differences are not always what they look like
RechenmannFrançoisThe algorithm we have presented works on an array of distance between sequences. These distances are evaluated on the basis of differences between the sequences. The problem is that behind the





Avec les mêmes intervenants et intervenantes
-
1.4. What is an algorithm?
RechenmannFrançoisWe have seen that a genomic textcan be indeed a very long sequence of characters. And to interpret this sequence of characters, we will need to use computers. Using computers means writing program.
-
2.2. Genes: from Mendel to molecular biology
RechenmannFrançoisThe notion of gene emerged withthe works of Gregor Mendel. Mendel studied the inheritance on some traits like the shape of pea plant seeds,through generations. He stated the famous laws of inheritance
-
2.10. How to find genes?
RechenmannFrançoisGetting the sequence of the genome is only the beginning, as I explained, once you have the sequence what you want to do is to locate the gene, to predict the function of the gene and maybe study the
-
3.8. Probabilistic methods
RechenmannFrançoisUp to now, to predict our gene,we only rely on the process of searching certain strings or patterns. In order to further improve our gene predictor, the idea is to use, to rely onprobabilistic methods
-
4.3. Measuring sequence similarity
RechenmannFrançoisSo we understand why gene orprotein sequences may be similar. It's because they evolve togetherwith the species and they evolve in time, there aremodifications in the sequence and that the sequence
-
5.4. The UPGMA algorithm
RechenmannFrançoisWe know how to fill an array with the values of the distances between sequences, pairs of sequences which are available in the file. This array of distances will be the input of our algorithm for
-
1.7. DNA walk
RechenmannFrançoisWe will now design a more graphical algorithm which is called "the DNA walk". We shall see what does it mean "DNA walk". Walk on to DNA. Something like that, yes. But first, just have a look again at
-
2.6. Algorithms + data structures = programs
RechenmannFrançoisBy writing the Lookup GeneticCode Function, we completed our translation algorithm. So we may ask the question about the algorithm, does it terminate? Andthe answer is yes, obviously. Is it pertinent,
-
3.3. Searching for start and stop codons
RechenmannFrançoisWe have written an algorithm for finding genes. But you remember that we arestill to write the two functions for finding the next stop codonand the next start codon. Let's see how we can do that. We
-
4.1. How to predict gene/protein functions?
RechenmannFrançoisLast week we have seen that annotating a genome means first locating the genes on the DNA sequences that is the genes, the region coding for proteins. But this is indeed the first step,the next very
-
4.10. How efficient is this algorithm?
RechenmannFrançoisWe have seen the principle of an iterative algorithm in two paths for aligning and comparing two sequences of characters, here DNA sequences. And we understoodwhy the iterative version is much more
-
1.2. At the heart of the cell: the DNA macromolecule
RechenmannFrançoisDuring the last session, we saw how at the heart of the cell there's DNA in the nucleus, sometimes of cells, or directly in the cytoplasm of the bacteria. The DNA is what we call a macromolecule, that










