Notice
5.4. L’algorithme UPGMA
- document 1 document 2 document 3
- niveau 1 niveau 2 niveau 3
Descriptif
L'algorithme, que nous allons étudier pour la reconstruction d'arbres phylogénétiques à partir des distances, s'appelle UPGMA. Un nom plutôt compliqué pour une méthode qui est plutôt simple. Et même, on le verra trop simple. UPGMA signifie Unweighted Pair Group Method with Arithmetic Mean. Nous allons voir au fur et à mesure, la signification dans l'exécution de l'algorithme de chacun de ces termes. Le point de départ de cet algorithme est donc un tableau de distances, tel que nous avons pu le remplir dans la session précédente. Voilà l'exemple que nous allons traiter. C'est un exemple simple. Nous avons sept espèces différentes et nous avons calculé les distances entre ces espèces à travers le calcul des distances, entre les séquences d'un gène homologue de ces espèces, à toutes ces espèces. Vous vous souvenez que le tableau que nous avons calculé était d'une part symétrique et que d'autre part, les valeurs sur la diagonale étaient sans surprise égales à 0. Ici nous avons choisi de ne conserver et de n'afficher que les valeurs significatives. Donc inutile de montrer les valeurs qui sont les symétriques des autres. Et inutile d'afficher les 0 sur les diagonales. Ce qui explique que notre tableau apparaît incomplet d'une certaine manière. La première étape de l'algorithme consiste à rechercher parmi toutes ces valeurs de distance dans le tableau la plus petite. Ici, c'est 2 et c'est la distance qui sépare l'espèce F de l'espèce C. Raccourci de langage, la distance qui sépare les séquences associées aux espèces F et C. C'est la distance la plus faible. Elle nous pousse donc à grouper ces 2 espèces dans un même sous-graphe en créant un noeud ancêtre ici. Ces 2 espèces sont proches, sont similaires parce qu'elles possèdent un ancêtre commun récent...
ERRATUM
Sur la slide 3 l’orateur parle de 7 espèces différentes, en fait il y en a 6.
Intervention


Dans la même collection
-
5.7. Les applications en microbiologie
RechenmannFrançoisParmentelatThierryUne très grande diversité, on l'a vu, d'algorithmes en bio-informatique, motivé par la résolution de problèmes différents. Ces algorithmes, ces recherches en bio-informatique, s'appuient sur des
-
5.1. L’arbre des espèces
RechenmannFrançoisParmentelatThierryDans cette cinquième et dernière partie de notre cours sur le génome et les algorithmes, qui se veut une introduction à l'analyse informatique de l'information génétique, nous regarderons de plus près
-
5.5. Quand les différences sont trompeuses
RechenmannFrançoisParmentelatThierryIl y a plusieurs raisons pour lesquelles la méthode UPGMA, que nous venons de voir, se révèle simpliste. L'une des raisons par exemple, c'est pourquoi quand on recalcule les distances, quand on a
-
5.2. L’arbre, objet abstrait
RechenmannFrançoisParmentelatThierryVous l'aurez compris un arbre phylogénétique est un arbre abstrait qui n'a qu'un lointain rapport métaphorique avec un véritable arbre. L'arbre des bio-informaticiens et des informaticiens se
-
5.6. La diversité des algorithmes informatiques
RechenmannFrançoisParmentelatThierryNous n'avons vu dans ce cours qu'un exemple extrêmement réduit d'algorithme bio informatique. Il existe en effet une très grande diversité de ces algorithmes bio informatiques qui sont motivés par l
-
5.3. Remplir un tableau de distances
RechenmannFrançoisParmentelatThierryPour tenter de construire l'arbre phylogénétique d'un ensemble d'espèces, nous allons utiliser les données et génotypique ou des données génotypiques disponibles sur ces espèces. Plus clairement, nous





Avec les mêmes intervenants et intervenantes
-
1.8. Compressing the DNA walk
RechenmannFrançoisWe have written the algorithm for the circle DNA walk. Just a precision here: the kind of drawing we get has nothing to do with the physical drawing of the DNA molecule. It is a symbolic
-
2.7. The algorithm design trade-off
RechenmannFrançoisWe saw how to increase the efficiencyof our algorithm through the introduction of a data structure. Now let's see if we can do even better. We had a table of index and weexplain how the use of these
-
3.4. Predicting all the genes in a sequence
RechenmannFrançoisWe have written an algorithm whichis able to locate potential genes on a sequence but only on one phase because we are looking triplets after triplets. Now remember that the genes maybe located on
-
4.7. Alignment costs
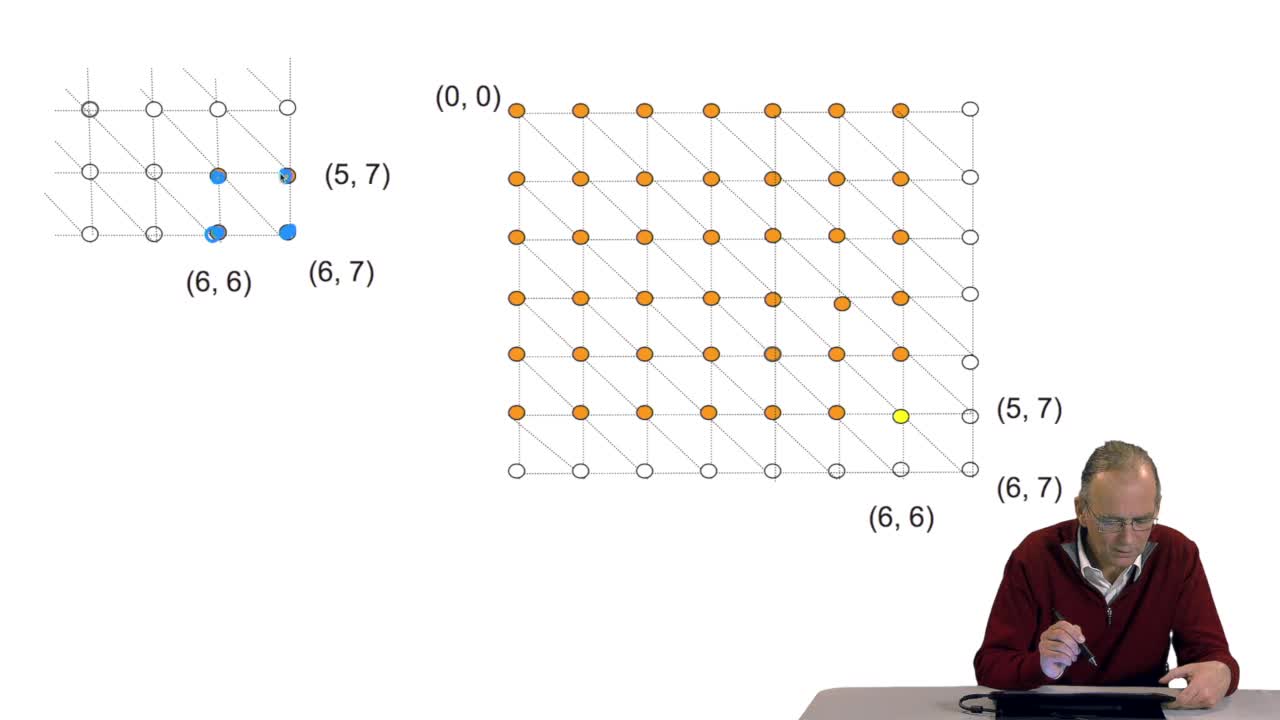
RechenmannFrançoisWe have seen how we can compute the cost of the path ending on the last node of our grid if we know the cost of the sub-path ending on the three adjacent nodes. It is time now to see more deeply why
-
4.9. Recursion can be avoided: an iterative version
RechenmannFrançoisWe have written a recursive function to compute the optimal path that is an optimal alignment between two sequences. Here all the examples I gave were onDNA sequences, four letter alphabet. OK. The
-
1.3. DNA codes for genetic information
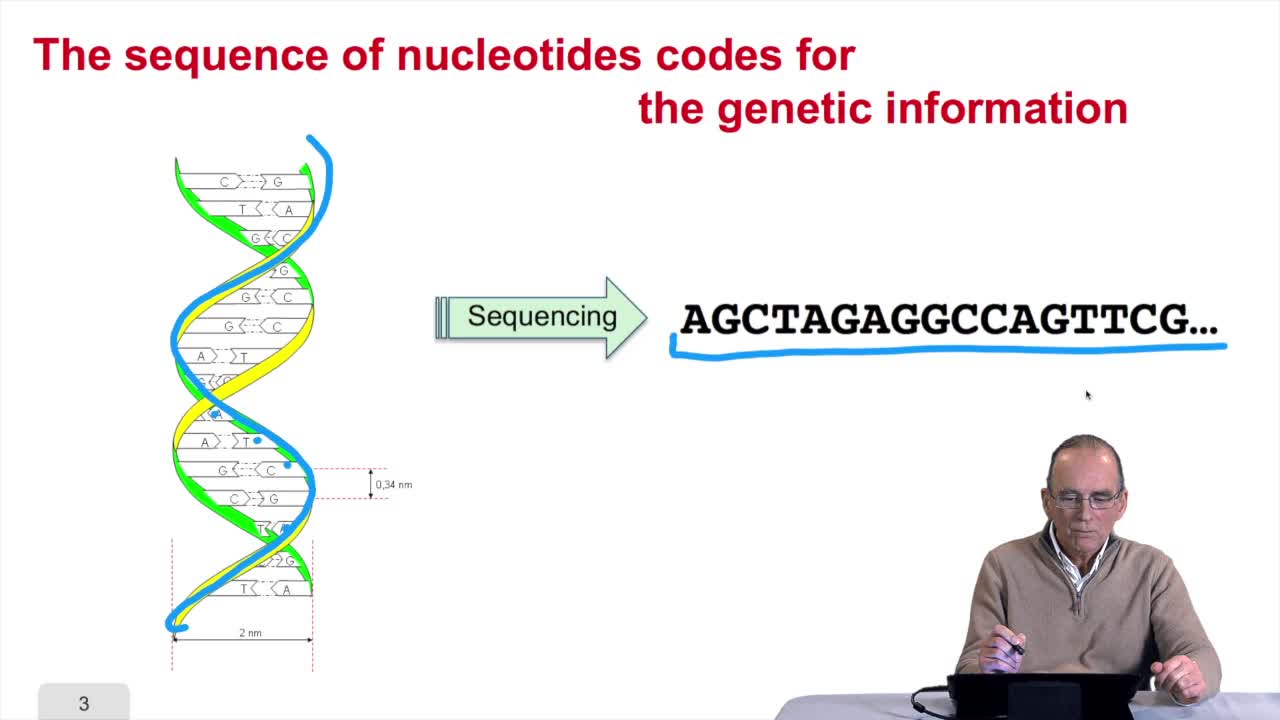
RechenmannFrançoisRemember at the heart of any cell,there is this very long molecule which is called a macromolecule for this reason, which is the DNA molecule. Now we will see that DNA molecules support what is called
-
2.1. The sequence as a model of DNA
RechenmannFrançoisWelcome back to our course on genomes and algorithms that is a computer analysis ofgenetic information. Last week we introduced the very basic concept in biology that is cell, DNA, genome, genes
-
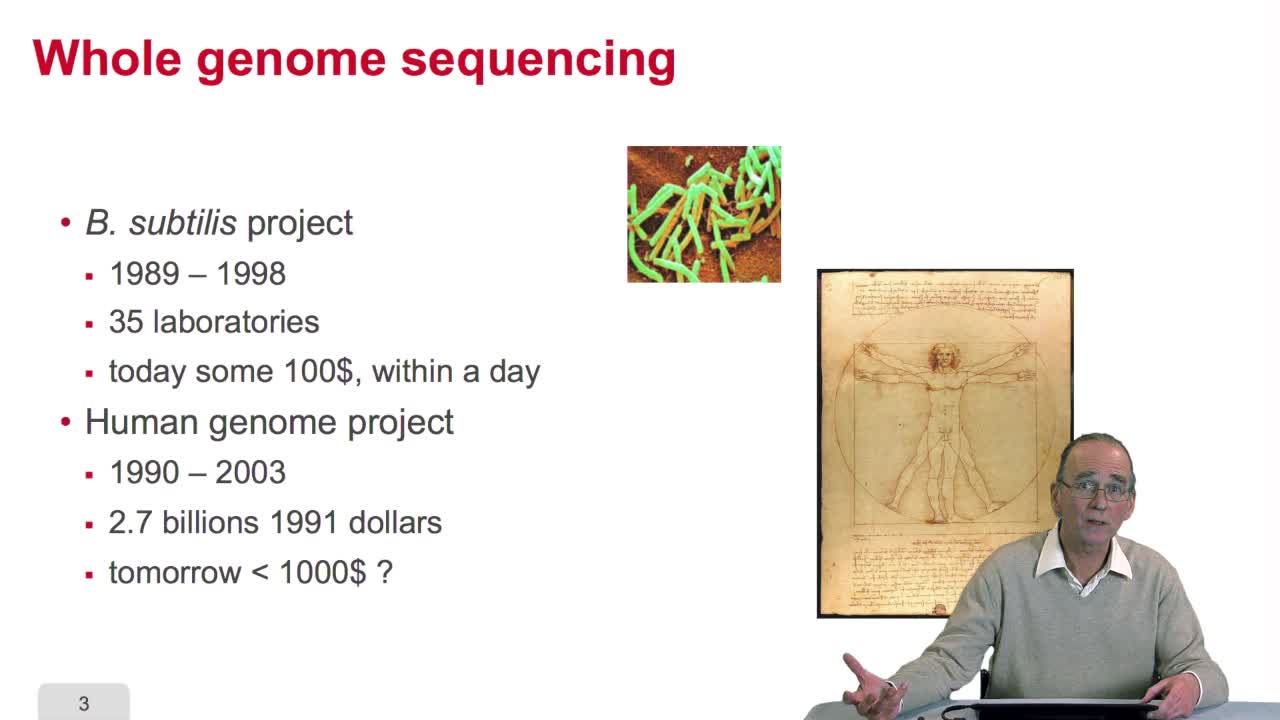
2.9. Whole genome sequencing
RechenmannFrançoisSequencing is anexponential technology. The progresses in this technologyallow now to a sequence whole genome, complete genome. What does it mean? Well let'stake two examples: some twenty years ago,
-
3.7. Index and suffix trees
RechenmannFrançoisWe have seen with the Boyer-Moore algorithm how we can increase the efficiency of spin searching through the pre-processing of the pattern to be searched. Now we will see that an alternative way of
-
4.4. Aligning sequences is an optimization problem
RechenmannFrançoisWe have seen a nice and a quitesimple solution for measuring the similarity between two sequences. It relied on the so-called hammingdistance that is counting the number of differencesbetween two
-
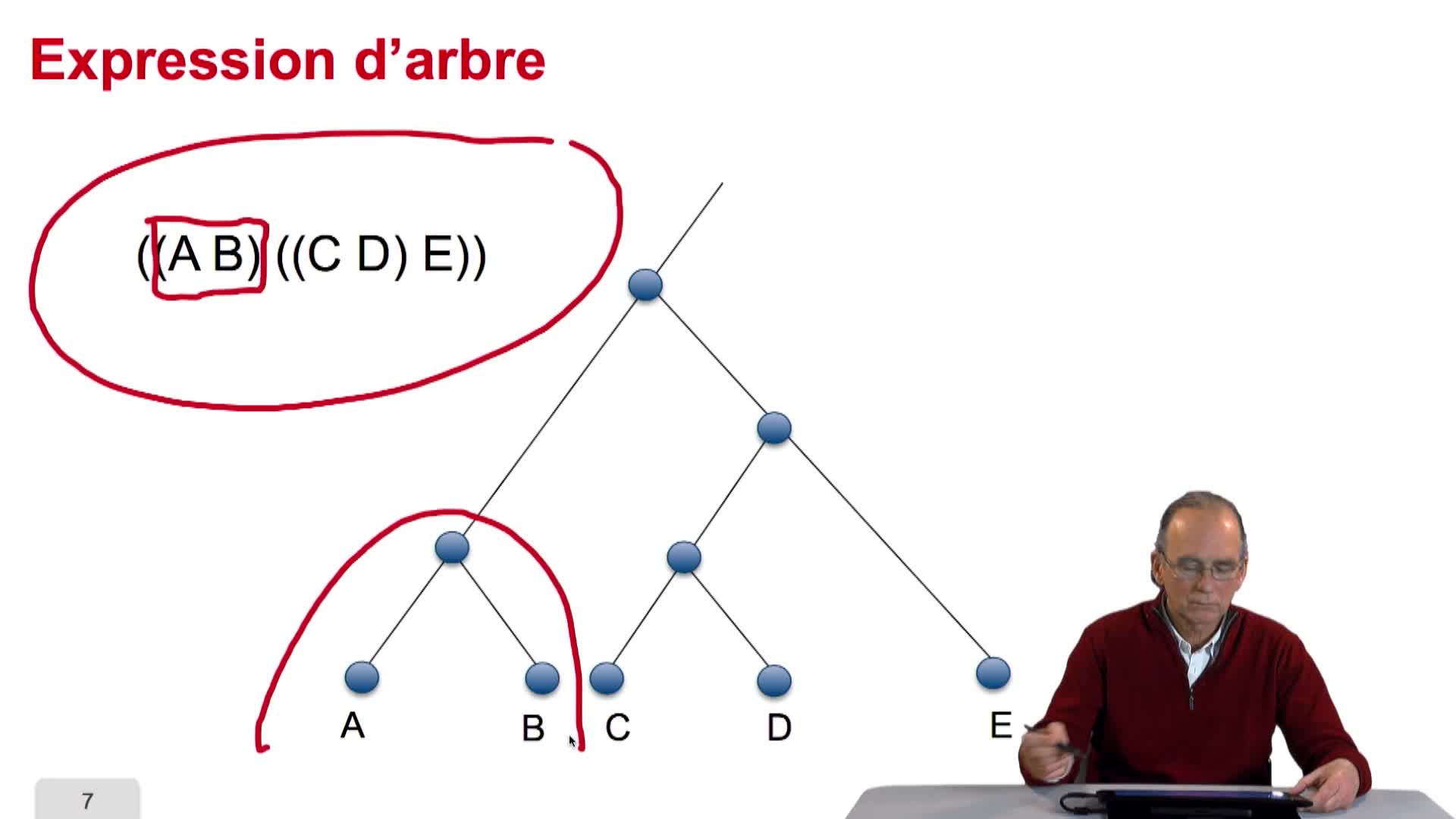
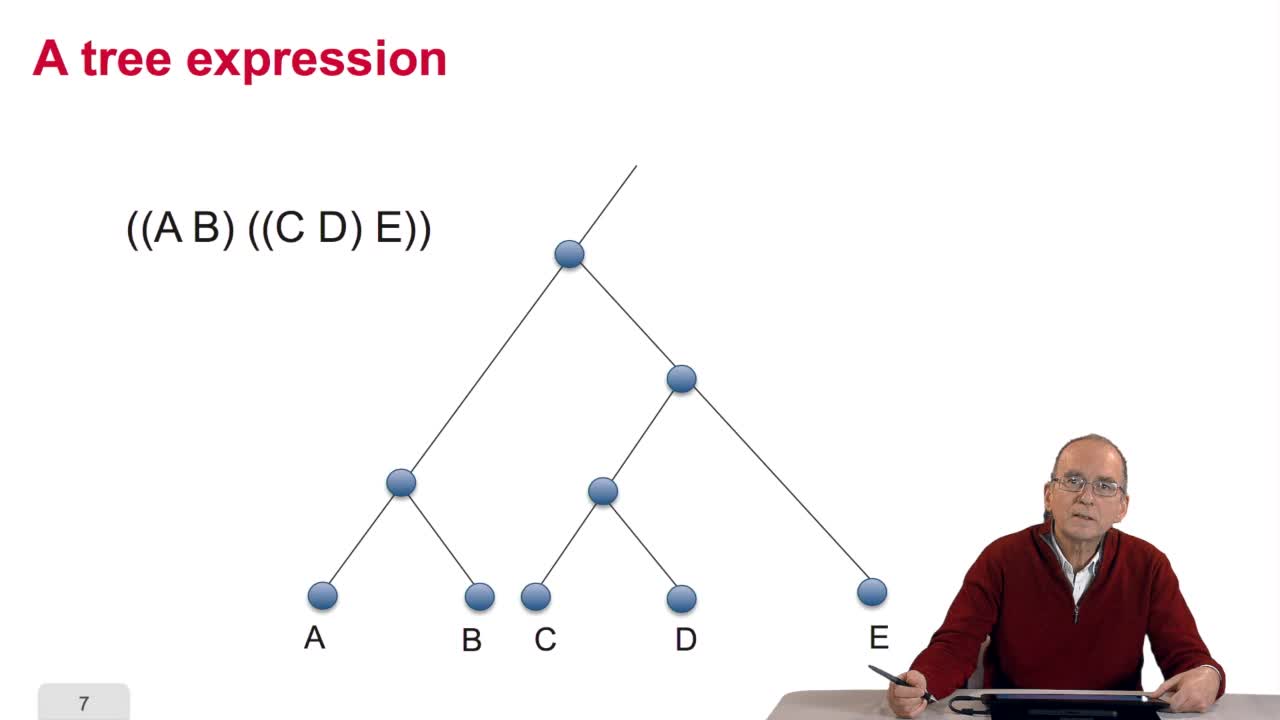
5.2. The tree, an abstract object
RechenmannFrançoisWhen we speak of trees, of species,of phylogenetic trees, of course, it's a metaphoric view of a real tree. Our trees are abstract objects. Here is a tree and the different components of this tree.
-
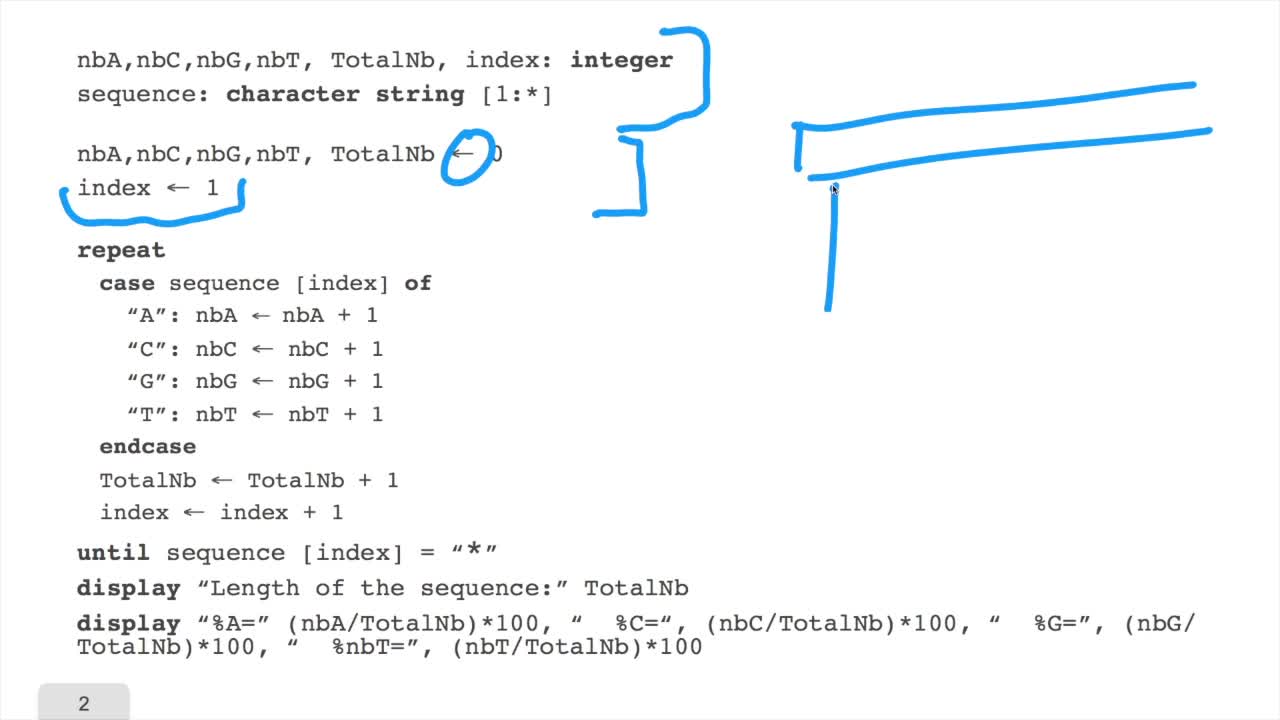
1.6. GC and AT contents of DNA sequence
RechenmannFrançoisWe have designed our first algorithmfor counting nucleotides. Remember, what we have writtenin pseudo code is first declaration of variables. We have several integer variables that are variables which
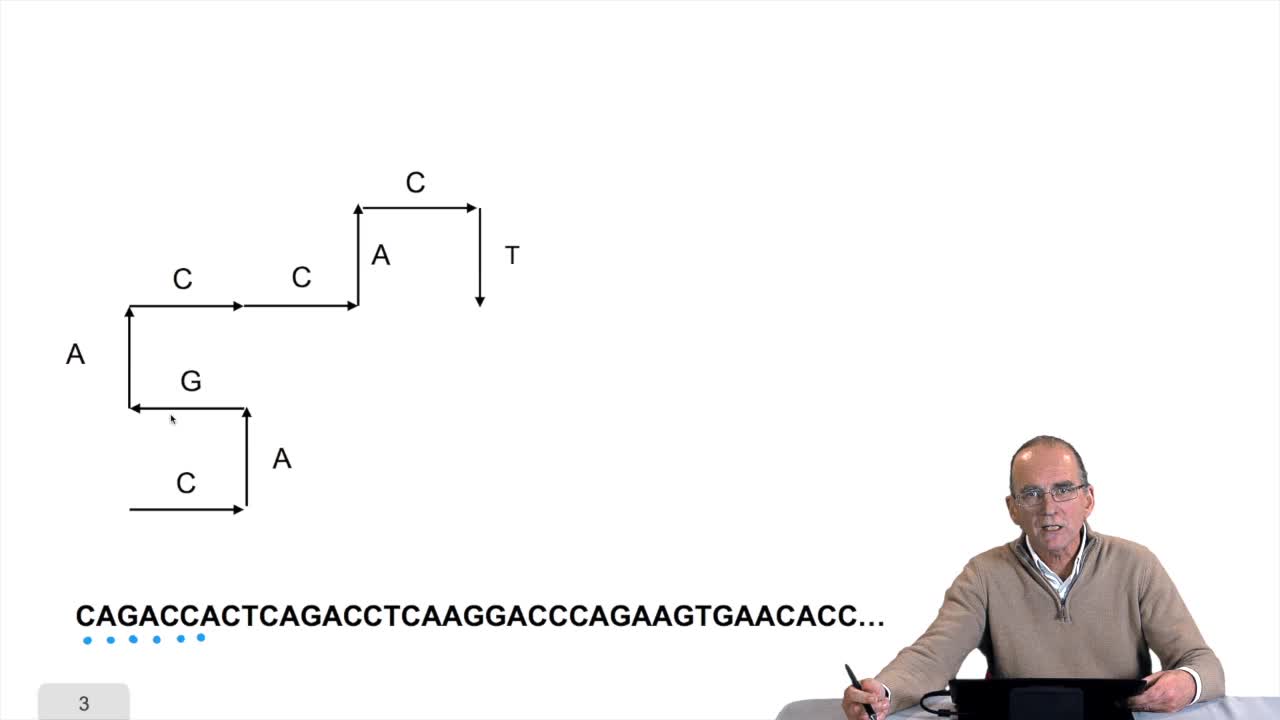






Sur le même thème
-
Machines algorithmiques, mythes et réalités
MazenodVincentVincent Mazenod, informaticien, partage le fruit de ses réflexions sur l'évolution des outils numériques, en lien avec les problématiques de souveraineté, de sécurité et de vie privée...
-
Désassemblons le numérique - #Episode11 : Les algorithmes façonnent-ils notre société ?
SchwartzArnaudLima PillaLaércioEstériePierreSalletFrédéricFerbosAudeRoumanosRayyaChraibi KadoudIkramUn an après le tout premier hackathon sur les méthodologies d'enquêtes journalistiques sur les algorithmes, ce nouvel épisode part à la rencontre de différents points de vue sur les algorithmes.
-
Les machines à enseigner. Du livre à l'IA...
BruillardÉricQue peut-on, que doit-on déléguer à des machines ? C'est l'une des questions explorées par Éric Bruillard qui, du livre aux IA génératives, expose l'évolution des machines à enseigner...
-
Désassemblons le numérique - #Episode9 : Bientôt des supercalculateurs dans nos piscines ?
BeaumontOlivierBouzelRémiDes supercalculateurs feraient-ils bientôt leur apparition dans les piscines municipales pour les chauffer ? Réponses d'Olivier Beaumont, responsable de l'équipe-projet Topal, et Rémi Bouzel,
-
Le projet dnarXiv : Stockage de données sur des molécules d'ADN
LavenierDominiqueDuprazElsaLeblancJulienCoatrieuxGouenouDominique Lavenier, Elsa Dupraz, Julien Leblanc et Gouenou Coatrieux nous présentent le projet dnarXiv, un projet porté par le LabEx CominLabs qui explore le stockage de données sur des molécules d
-
Projection methods for community detection in complex networks
LitvakNellyCommunity detection is one of most prominent tasks in the analysis of complex networks such as social networks, biological networks, and the world wide web. A community is loosely defined as a group
-
Lara Croft. doing fieldwork under surveillance
Dall'AgnolaJasminLara Croft. Doing Fieldwork Under Surveillance Intervention de Jasmin Dall'Agnola (The George Washington University), dans le cadre du Colloque coorganisé par Anders Albrechtslund, professeur en
-
Containing predictive tokens in the EU
CzarnockiJanContaining Predictive Tokens in the EU – Mapping the Laws Against Digital Surveillance, intervention de Jan Czarnocki (KU Leuven), dans le cadre du Colloque coorganisé par Anders Albrechtslund,
-
Ivan Murit - Processus de création d'images
MuritIvanJe vais présenter une manière décalée d'aborder les outils d'impression. Pour cela nous ne partirons pas de l'envie d'imprimer une image préexistante, mais d'avant cela : comment se crée une forme
-
Le Creativ’Lab, au cœur de la robotique et de l’intelligence artificielle (ASR N°18 - LORIA)
HénaffPatrickLefebvreSylvainLe LORIA, laboratoire phare de la Grande Région dans le domaine de l’informatique, propose de rendre la recherche plus ouverte, plus collaborative, plus ambitieuse… en un mot, plus créative, à travers
-
Les algorithmes de Parcoursup
MathieuClaireL’objectif de la journée « Algorithmes d’aide à la décision publique » était de sensibiliser le grand public aux rôles des algorithmes d’aide à la décision publique utilisés par exemple pour l
-
Algorithmes d'aide à la décision publique / Ouverture
RéveillèreLaurentMaveyraud-TricoireSamuelBlancXavierBertrandYvesMainguenéMarcL’objectif de la journée « Algorithmes d’aide à la décision publique » était de sensibiliser le grand public aux rôles des algorithmes d’aide à la décision publique utilisés par exemple pour l











