Notice
4.2. Évolution et similarité de séquences
- document 1 document 2 document 3
- niveau 1 niveau 2 niveau 3
Descriptif
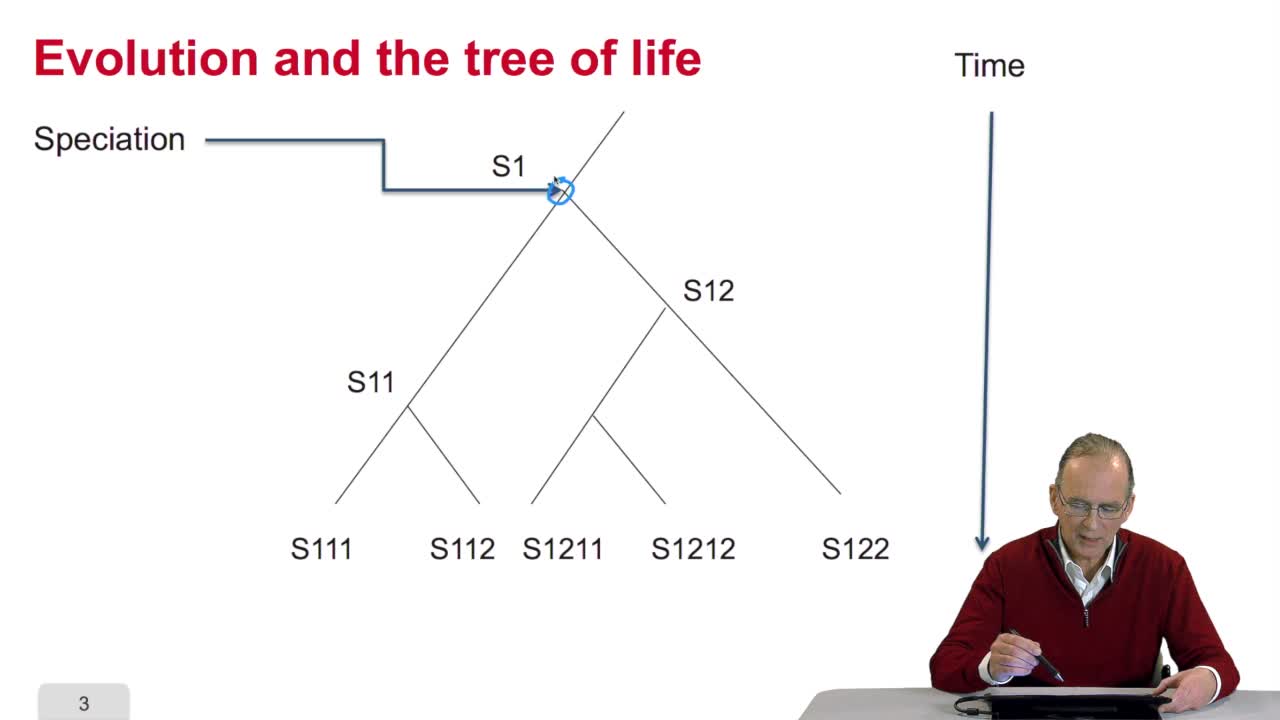
Avant de chercher à quantifier ce qu'est la similarité de séquence, on peut se poser la question même de savoir pourquoi des séquences de génome sont similaires entre organismes. La réponse tient dans la théorie de l'évolution que l'on doit à Charles Darwin. Que dit Charles Darwin et que disent les biologistes évolutionnistes actuellement ? Ils disent que les espèces évoluent. Une espèce, par spéciation, donne naissance à 2 autres espèces qui évoluent et ainsi de suite. D'où cet arbre du vivant qu'esquissait déjà Darwin, grand penseur, dans ses carnets.
En pratique, que cela signifie-t-il ? On peut figurer l'arbre des espèces sous la forme d'un arbre informatique, on y reviendra la semaine prochaine, où figurent aux nœuds, les espèces. Ici existe une espèce qui, par ce processus dit de spéciation, évolue sous la forme de 2 espèces distinctes. Celle-ci évolue encore sous forme de 2 espèces distinctes et ainsi de suite. Le temps figure là puisque cette espèce-là existe à un moment donné, donne lieu à 2 autres espèces de façon ultérieure et ainsi de suite...
Intervention / Responsable scientifique


Dans la même collection
-
4.8. Un algorithme récursif
RechenmannFrançoisParmentelatThierryNous avons désormais en main tous les éléments pour écrire notre algorithme de détermination d'un alignement optimal, ici d'un chemin optimal. Avec les notations que nous avons introduites, je vous
-
4.3. Quantifier la similarité de deux séquences
RechenmannFrançoisParmentelatThierryLe principe est donc de rechercher, dans les bases de données, des séquences similaires à celles que nous sommes en train d'étudier. Nous faisons aussi l'hypothèse que plus les séquences sont
-
4.6. Si un chemin est optimal, tous ses chemins partiels sont optimaux
RechenmannFrançoisParmentelatThierryNous cherchons à concevoir un algorithme capable de déterminer l'alignement optimal de 2 séquences. Et nous avons vu que ça revient à chercher un algorithme qui recherche un chemin optimal dans une
-
4.9. Éviter la récursivité : une version itérative
RechenmannFrançoisParmentelatThierryLa fonction récursive que nous avons obtenue est d'un code assez compact et plutôt élégant, mais effectivement peu efficace. Pourquoi ? Rappelons son fonctionnement. Cette fonction est d'abord appelée
-
4.4. L’alignement de séquences devient un problème d’optimisation
RechenmannFrançoisParmentelatThierryLa distance de Hamming nous donne une première possibilité de mesurer la similarité entre 2 séquences. Mais elle ne reflète pas suffisamment la réalité biologique. Qu'est-ce que j'entends par là ? On
-
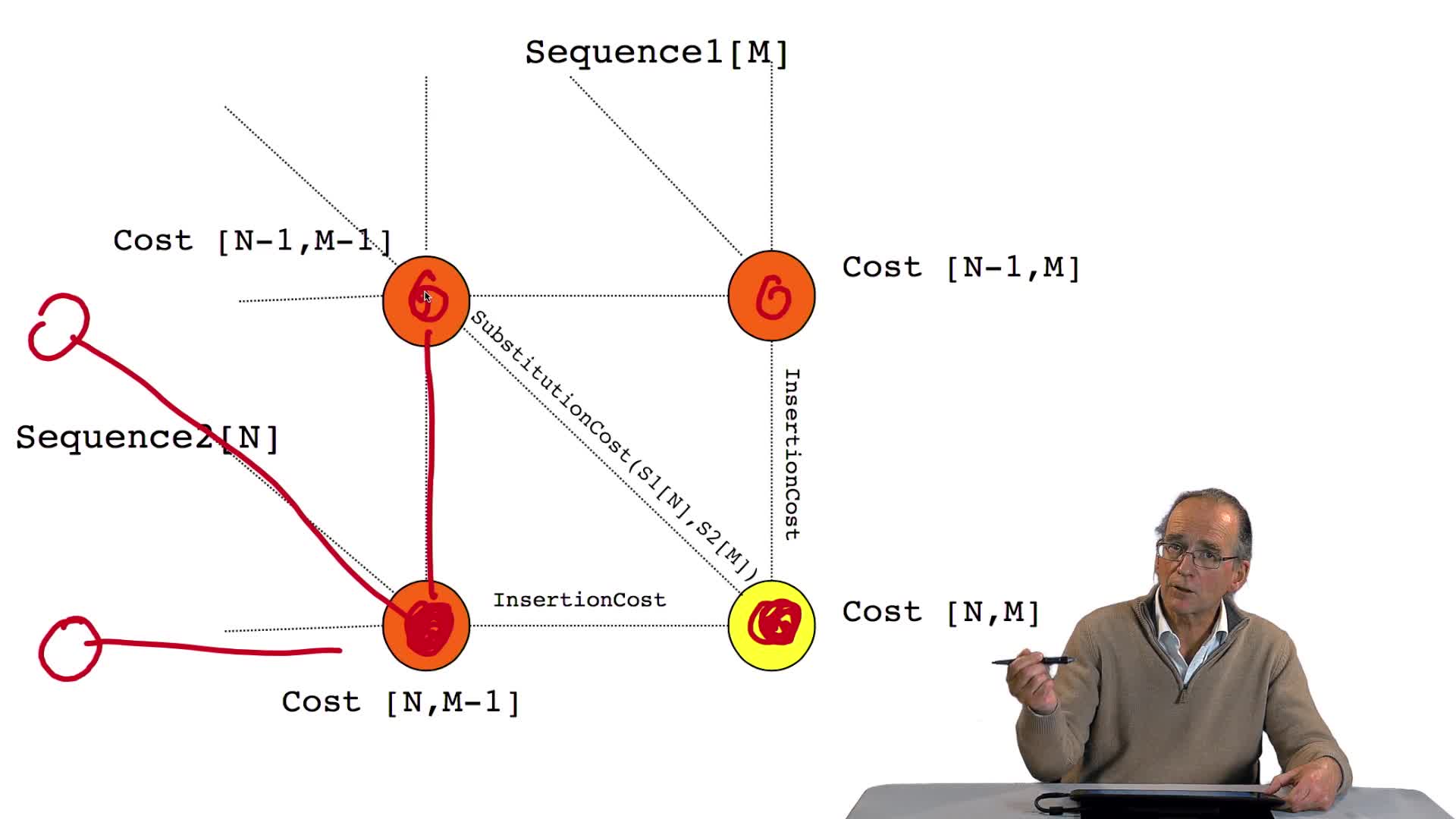
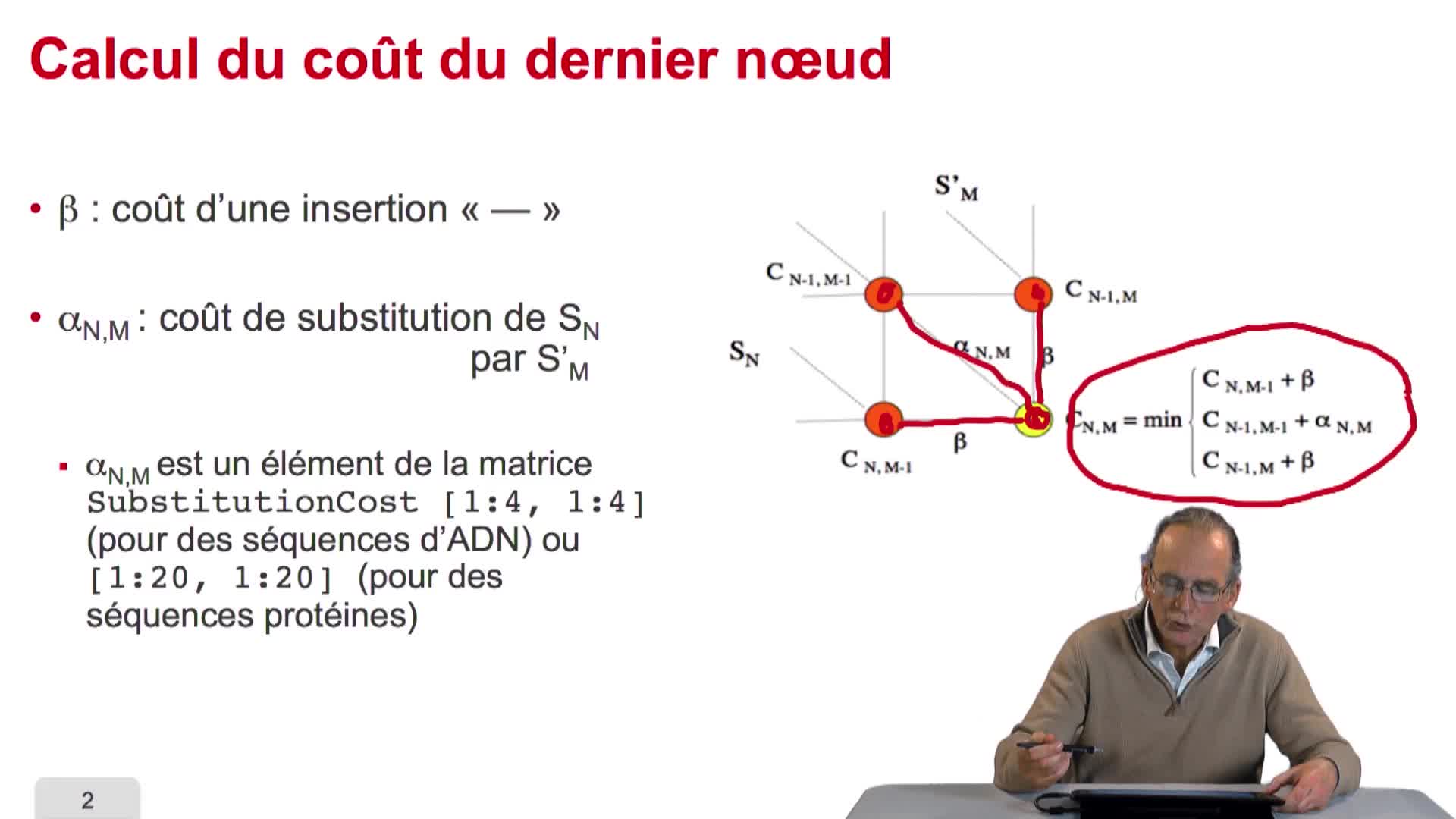
4.7. Coûts et alignement
RechenmannFrançoisParmentelatThierryNous avons vu l'ébauche de notre algorithme d'alignement optimal en considérant la possibilité de calculer le coût optimal, ou score optimal, de ce dernier noeud. Et nous avons vu que le coût de ce
-
4.1. Comment prédire les fonctions des gènes/protéines ?
RechenmannFrançoisParmentelatThierryAprès avoir regardé dans les yeux, les semaines précédentes, l'ADN, vu comment cet ADN par séquençage produisait des textes, des séquences génomiques, étudié la relation entre gènes et protéines,
-
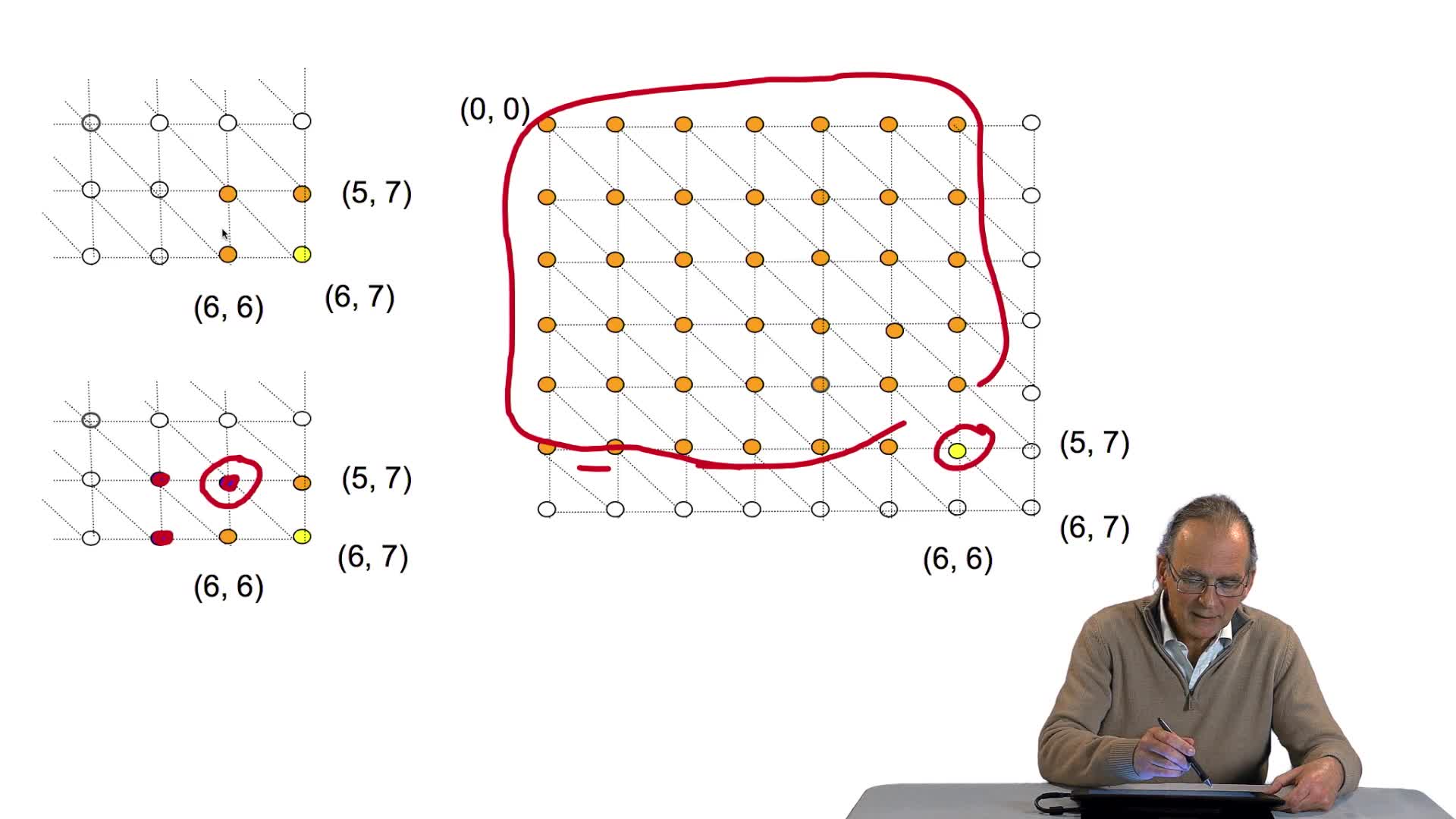
4.10. Cet algorithme est-il efficace ?
RechenmannFrançoisParmentelatThierryLa version itérative de notre algorithme d'alignement optimal de séquences est indéniablement beaucoup plus efficace que sa version récursive, puisque nous avons vu qu'il permettait d'éviter que le
-
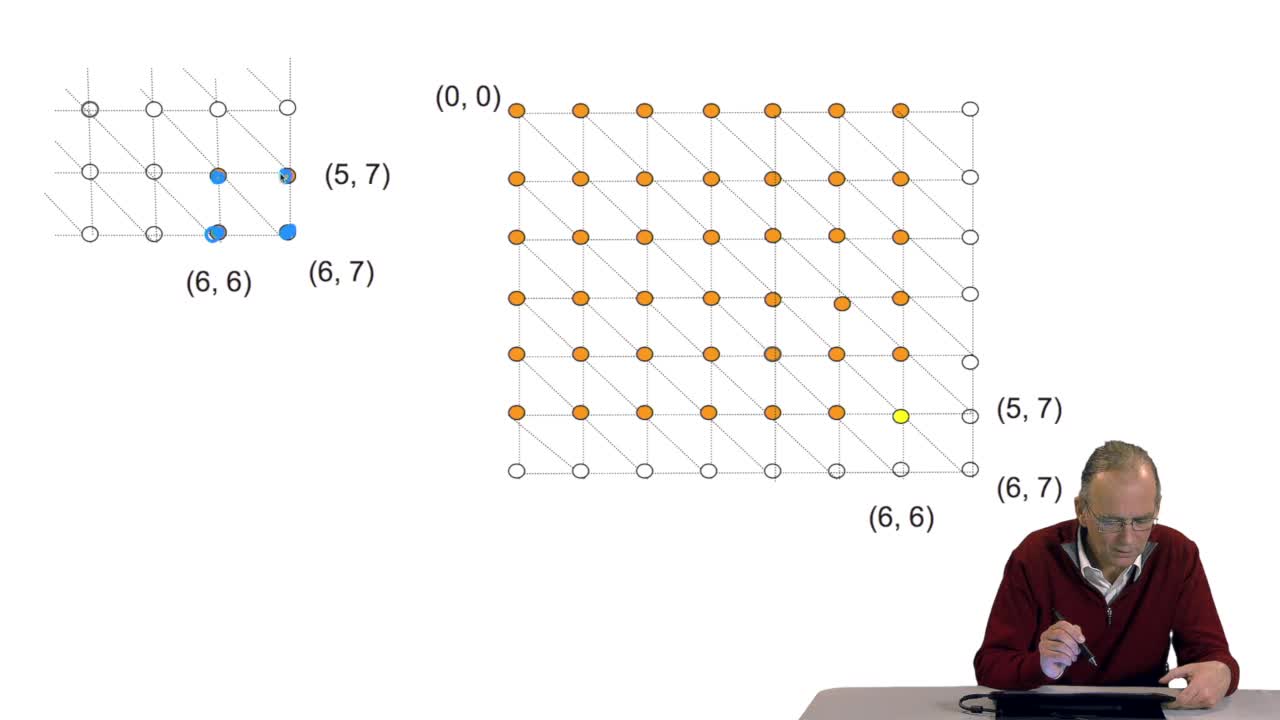
4.5. Un alignement de séquences vu comme un chemin dans une grille
RechenmannFrançoisParmentelatThierryPour comparer deux séquences entre elles, il faut donc les aligner. Aligner ces deux séquences suppose faire des hypothèses d'insertion, délétion, aux bons endroits. Ça signifie, d'un point de vue






Avec les mêmes intervenants et intervenantes
-
1.5. Counting nucleotides
RechenmannFrançoisIn this session, don't panic. We will design our first algorithm. This algorithm is forcounting nucleotides. The idea here is that as an input,you have a sequence of nucleotides, of bases, of letters,
-
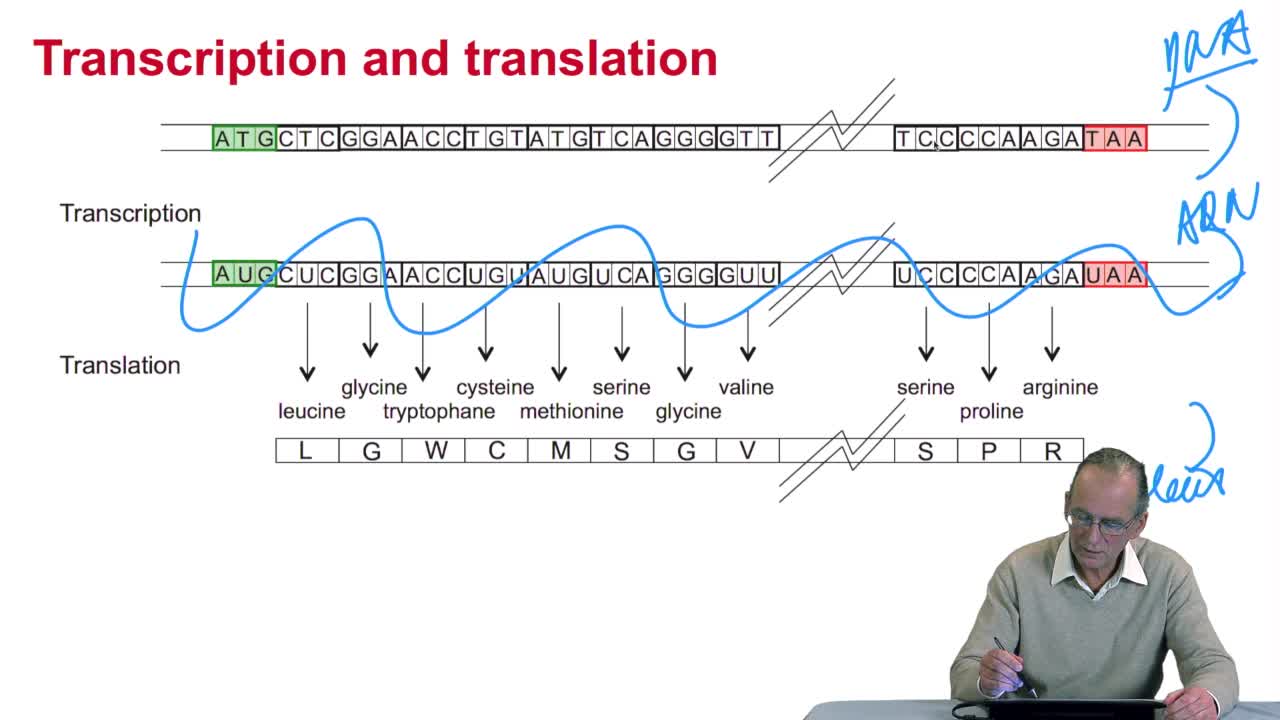
2.4. A translation algorithm
RechenmannFrançoisWe have seen that the genetic codeis a correspondence between the DNA or RNA sequences and aminoacid sequences that is proteins. Our aim here is to design atranslation algorithm, we make the
-
3.1. All genes end on a stop codon
RechenmannFrançoisLast week we studied genes and proteins and so how genes, portions of DNA, are translated into proteins. We also saw the very fast evolutionof the sequencing technology which allows for producing
-
3.9. Benchmarking the prediction methods
RechenmannFrançoisIt is necessary to underline that gene predictors produce predictions. Predictions mean that you have no guarantees that the coding sequences, the coding regions,the genes you get when applying your
-
4.2. Why gene/protein sequences may be similar?
RechenmannFrançoisBefore measuring the similaritybetween the sequences, it's interesting to answer the question: why gene or protein sequences may be similar? It is indeed veryinteresting because the answer is related
-
5.4. The UPGMA algorithm
RechenmannFrançoisWe know how to fill an array with the values of the distances between sequences, pairs of sequences which are available in the file. This array of distances will be the input of our algorithm for
-
1.8. Compressing the DNA walk
RechenmannFrançoisWe have written the algorithm for the circle DNA walk. Just a precision here: the kind of drawing we get has nothing to do with the physical drawing of the DNA molecule. It is a symbolic
-
2.7. The algorithm design trade-off
RechenmannFrançoisWe saw how to increase the efficiencyof our algorithm through the introduction of a data structure. Now let's see if we can do even better. We had a table of index and weexplain how the use of these
-
3.4. Predicting all the genes in a sequence
RechenmannFrançoisWe have written an algorithm whichis able to locate potential genes on a sequence but only on one phase because we are looking triplets after triplets. Now remember that the genes maybe located on
-
4.7. Alignment costs
RechenmannFrançoisWe have seen how we can compute the cost of the path ending on the last node of our grid if we know the cost of the sub-path ending on the three adjacent nodes. It is time now to see more deeply why
-
4.9. Recursion can be avoided: an iterative version
RechenmannFrançoisWe have written a recursive function to compute the optimal path that is an optimal alignment between two sequences. Here all the examples I gave were onDNA sequences, four letter alphabet. OK. The
-
1.3. DNA codes for genetic information
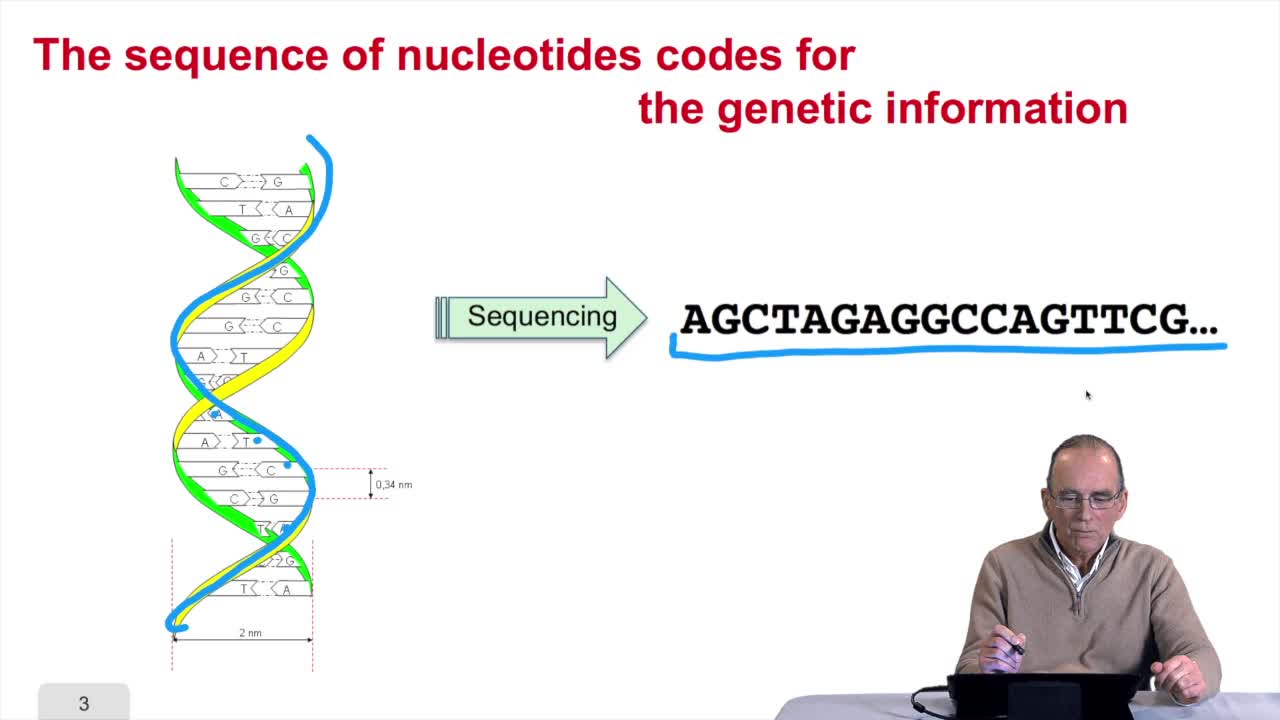
RechenmannFrançoisRemember at the heart of any cell,there is this very long molecule which is called a macromolecule for this reason, which is the DNA molecule. Now we will see that DNA molecules support what is called







Sur le même thème
-
La voix, une donnée identifiante à protéger
VincentEmmanuelEmmanuel Vincent, chercheur au Centre Inria de l'Université de Lorraine et au Loria (Laboratoire lorrain de recherche en informatique et ses applications), présente sa recherche sur l'anonymisation de
-
Podcast 1/4 d'heure avec : Emmanuel Vincent, chercheur au Centre Inria de l'Université de Lorraine …
VincentEmmanuelRencontre avec Emmanuel Vincent - chercheur au Centre Inria de l'Université de Lorraine et Loria (Laboratoire lorrain de recherche en informatique et ses applications).
-
Stockage de données numériques sur ADN synthétique : Introduction au domaine
AntoniniMarcDuprazElsaLavenierDominiquePrésentation globale des différentes étapes du stockage de données sur des molécules d'ADN synthétique
-
Stockage de données numériques sur ADN synthétique : Production des données: synthèse, séquençage
LavenierDominiqueBarbryPascalDescription des opérations d'écriture et de lecture des molécules d'ADN : synthèse et séquençage.
-
Stockage de données numériques sur ADN synthétique : Reconstruction des données
LavenierDominiqueTraitement des données après séquençage
-
Stockage de données numériques sur ADN synthétique : Codage Canal
DuprazElsaTechniques de codage pour le stockage de données sur ADN
-
Stockage de données numériques sur ADN synthétique : Codage Source
AntoniniMarcCodage source pour le stockage de données sur ADN synthétique
-
Stockage de données numériques sur ADN synthétique : Théorie de l'information
Kas HannaSergeQuelle quantité d'information peut-on stocker et récupérer de manière fiable dans l'ADN ?
-
The tree of life
AbbySophieLes Rencontres Exobiologiques pour Doctorants (RED) sont une école de formation sur les « bases de l'astrobiologie ». L’édition 2025 s’est tenue du 16 au 21 mars au Parc Ornithologique du Teich.
-
Machines algorithmiques, mythes et réalités
MazenodVincentVincent Mazenod, informaticien, partage le fruit de ses réflexions sur l'évolution des outils numériques, en lien avec les problématiques de souveraineté, de sécurité et de vie privée...
-
Désassemblons le numérique - #Episode11 : Les algorithmes façonnent-ils notre société ?
SchwartzArnaudLima PillaLaércioEstériePierreSalletFrédéricFerbosAudeRoumanosRayyaChraibi KadoudIkramUn an après le tout premier hackathon sur les méthodologies d'enquêtes journalistiques sur les algorithmes, ce nouvel épisode part à la rencontre de différents points de vue sur les algorithmes.
-
Les machines à enseigner. Du livre à l'IA...
BruillardÉricQue peut-on, que doit-on déléguer à des machines ? C'est l'une des questions explorées par Éric Bruillard qui, du livre aux IA génératives, expose l'évolution des machines à enseigner...











