Notice
5.2. L’arbre, objet abstrait
- document 1 document 2 document 3
- niveau 1 niveau 2 niveau 3
Descriptif
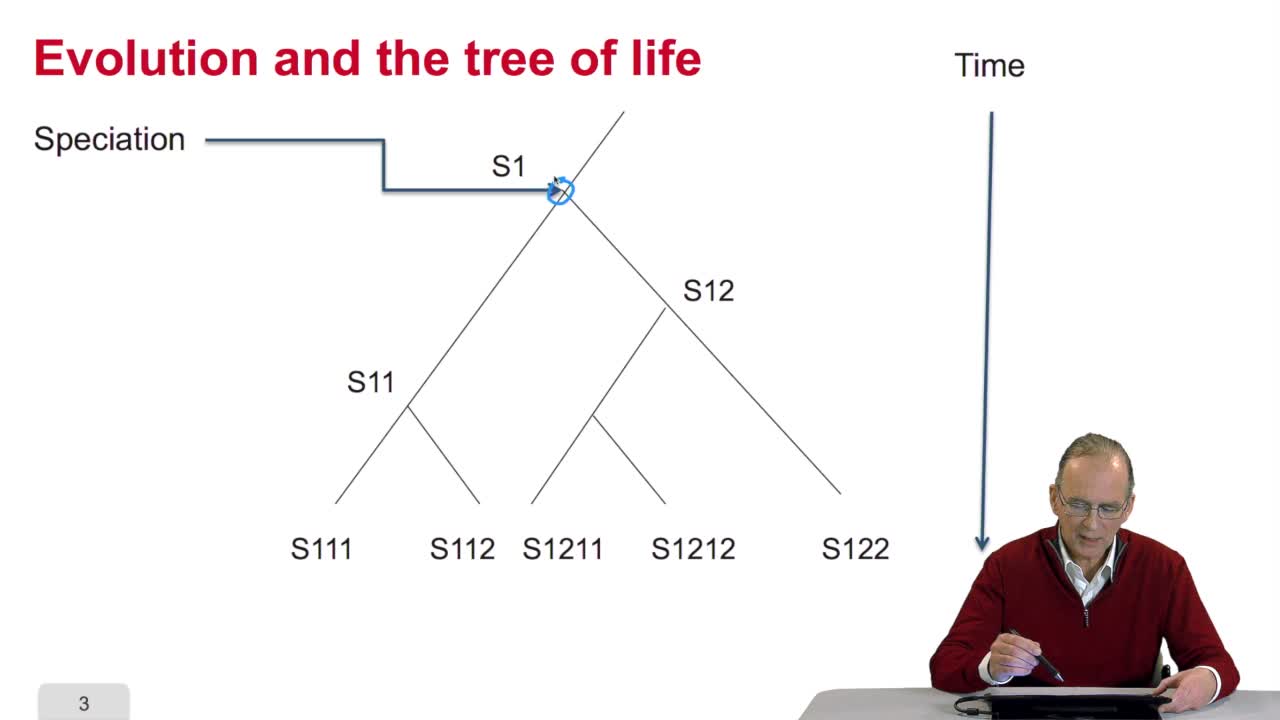
Vous l'aurez compris un arbre phylogénétique est un arbre abstrait qui n'a qu'un lointain rapport métaphorique avec un véritable arbre. L'arbre des bio-informaticiens et des informaticiens se dessinent du reste dans l'autre sens. C'est-à-dire que si on retrouve bien effectivement des branches qui connectent des noeuds, on a un noeud qui est la racine et qui est situé tout en haut et on situe en bas généralement par convention, les feuilles qu'on appelle également noeuds terminaux. Pour décrire un arbre, on peut utiliser une expression parenthésée, dont la logique s'impose assez rapidement. Voilà ici l'expression parenthésée correspondant à cette structure d'arbres. On voit cette sous-expression correspondre à ce sous-arbre. Très logiquement, cette expression correspond à ce sous-arbre. Et donc, cette expression-là correspond au sous-arbre incluant celui-ci, plus le noeud E. Et l'arbre tout entier est de noter par cette expression. Nous avons donc là un moyen non ambigu de noter une topologie, une structure d'arbre. Bien entendu, cette topologie d'arbres ne change pas suivant la manière dont on le dessine. On peut également avoir des expressions parenthésées différentes mais qui sont équivalentes du point de vue descriptif. Cette expression parenthésée est strictement équivalente à celle-ci. Elle décrit la même structure d'arbres. De même, les arbres peuvent prendre plusieurs apparences, suivant le style de dessin utilisé, la topologie reste la même. Cette manière de représenter les arbres est de loin préférée par les biologistes...
Intervention / Responsable scientifique


Dans la même collection
-
5.5. Quand les différences sont trompeuses
RechenmannFrançoisParmentelatThierryIl y a plusieurs raisons pour lesquelles la méthode UPGMA, que nous venons de voir, se révèle simpliste. L'une des raisons par exemple, c'est pourquoi quand on recalcule les distances, quand on a
-
5.3. Remplir un tableau de distances
RechenmannFrançoisParmentelatThierryPour tenter de construire l'arbre phylogénétique d'un ensemble d'espèces, nous allons utiliser les données et génotypique ou des données génotypiques disponibles sur ces espèces. Plus clairement, nous
-
5.6. La diversité des algorithmes informatiques
RechenmannFrançoisParmentelatThierryNous n'avons vu dans ce cours qu'un exemple extrêmement réduit d'algorithme bio informatique. Il existe en effet une très grande diversité de ces algorithmes bio informatiques qui sont motivés par l
-
5.4. L’algorithme UPGMA
RechenmannFrançoisParmentelatThierryL'algorithme, que nous allons étudier pour la reconstruction d'arbres phylogénétiques à partir des distances, s'appelle UPGMA. Un nom plutôt compliqué pour une méthode qui est plutôt simple. Et même,
-
5.7. Les applications en microbiologie
RechenmannFrançoisParmentelatThierryUne très grande diversité, on l'a vu, d'algorithmes en bio-informatique, motivé par la résolution de problèmes différents. Ces algorithmes, ces recherches en bio-informatique, s'appuient sur des
-
5.1. L’arbre des espèces
RechenmannFrançoisParmentelatThierryDans cette cinquième et dernière partie de notre cours sur le génome et les algorithmes, qui se veut une introduction à l'analyse informatique de l'information génétique, nous regarderons de plus près






Avec les mêmes intervenants et intervenantes
-
1.2. At the heart of the cell: the DNA macromolecule
RechenmannFrançoisDuring the last session, we saw how at the heart of the cell there's DNA in the nucleus, sometimes of cells, or directly in the cytoplasm of the bacteria. The DNA is what we call a macromolecule, that
-
1.10. Overlapping sliding window
RechenmannFrançoisWe have made some drawings along a genomic sequence. And we have seen that although the algorithm is quite simple, even if some points of the algorithmare bit trickier than the others, we were able to
-
2.3. The genetic code
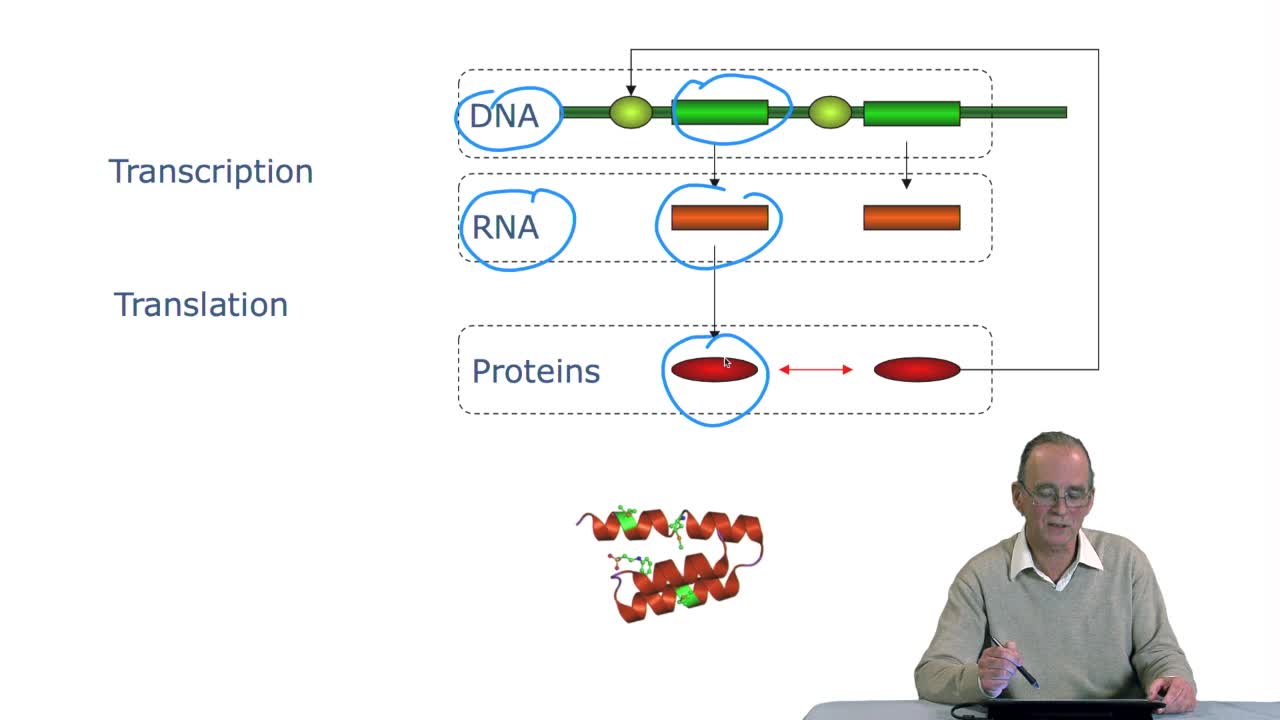
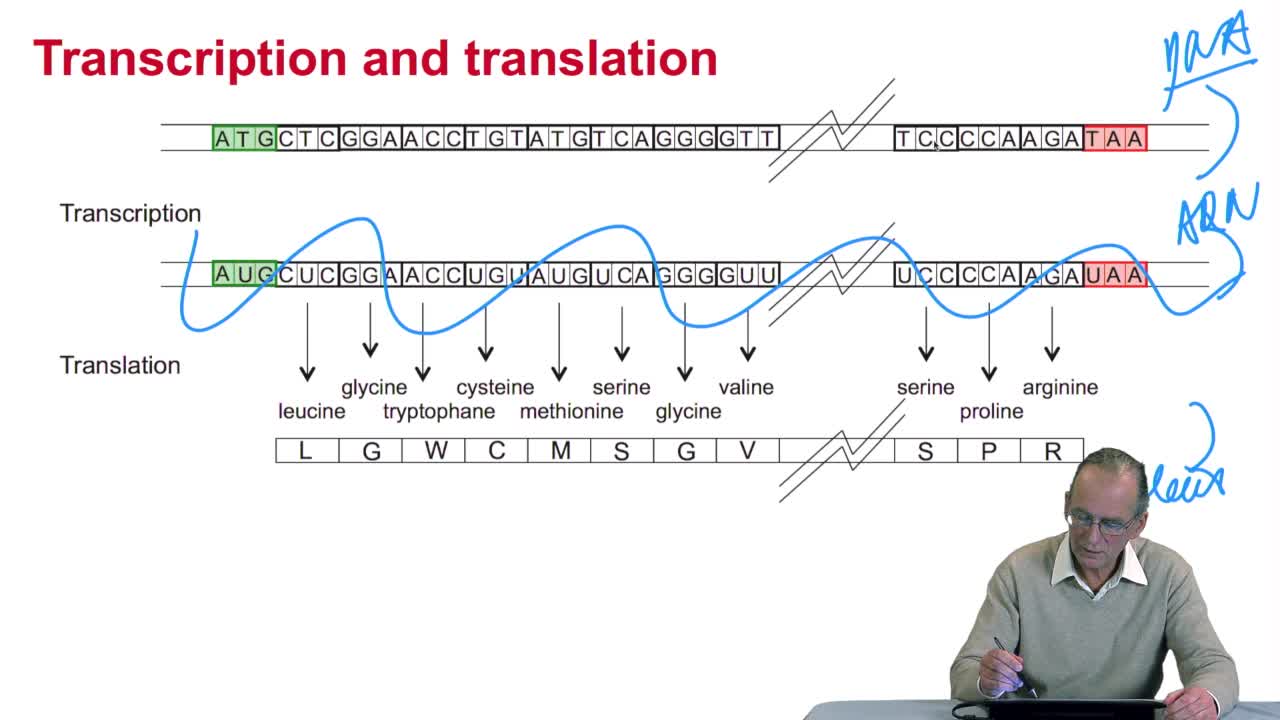
RechenmannFrançoisGenes code for proteins. What is the correspondence betweenthe genes, DNA sequences, and the structure of proteins? The correspondence isthe genetic code. Proteins have indeedsequences of amino acids.
-
3.6. Boyer-Moore algorithm
RechenmannFrançoisWe have seen how we can make gene predictions more reliable through searching for all the patterns,all the occurrences of patterns. We have seen, for example, howif we locate the RBS, Ribosome
-
4.5. A sequence alignment as a path
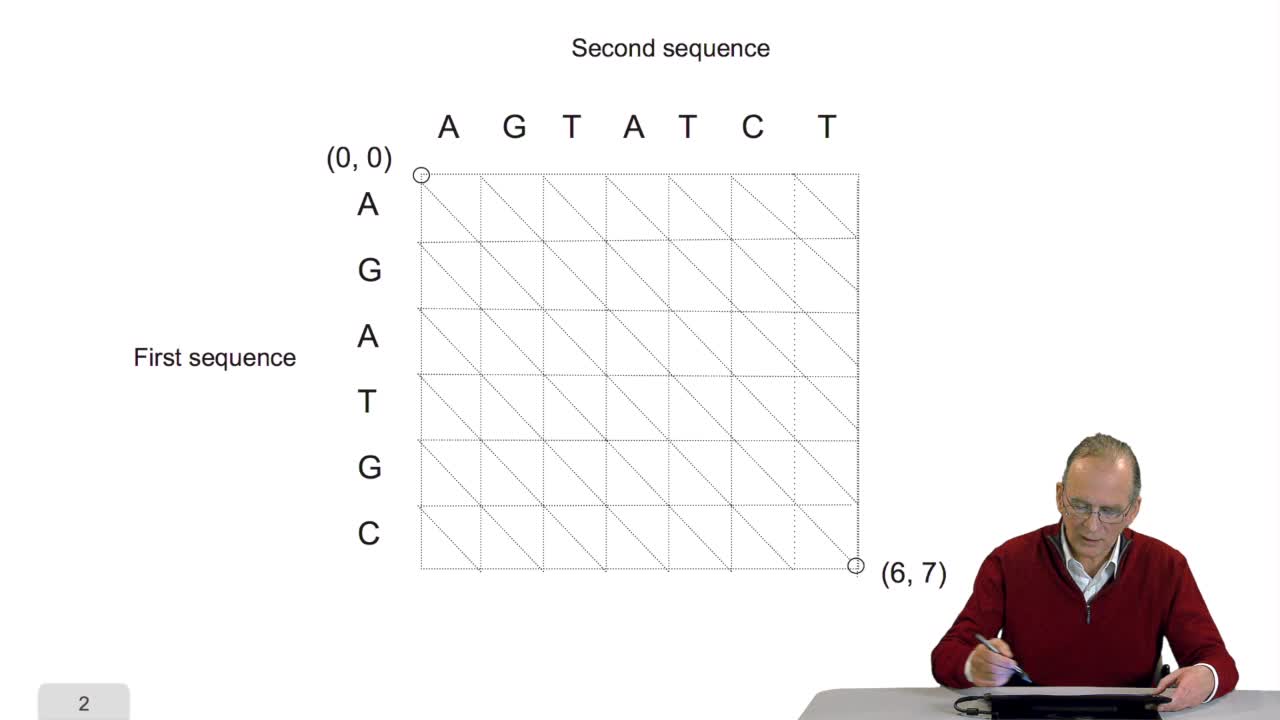
RechenmannFrançoisComparing two sequences and thenmeasuring their similarities is an optimization problem. Why? Because we have seen thatwe have to take into account substitution and deletion. During the alignment, the
-
5.5. Differences are not always what they look like
RechenmannFrançoisThe algorithm we have presented works on an array of distance between sequences. These distances are evaluated on the basis of differences between the sequences. The problem is that behind the
-
1.5. Counting nucleotides
RechenmannFrançoisIn this session, don't panic. We will design our first algorithm. This algorithm is forcounting nucleotides. The idea here is that as an input,you have a sequence of nucleotides, of bases, of letters,
-
2.4. A translation algorithm
RechenmannFrançoisWe have seen that the genetic codeis a correspondence between the DNA or RNA sequences and aminoacid sequences that is proteins. Our aim here is to design atranslation algorithm, we make the
-
3.1. All genes end on a stop codon
RechenmannFrançoisLast week we studied genes and proteins and so how genes, portions of DNA, are translated into proteins. We also saw the very fast evolutionof the sequencing technology which allows for producing
-
3.9. Benchmarking the prediction methods
RechenmannFrançoisIt is necessary to underline that gene predictors produce predictions. Predictions mean that you have no guarantees that the coding sequences, the coding regions,the genes you get when applying your
-
4.2. Why gene/protein sequences may be similar?
RechenmannFrançoisBefore measuring the similaritybetween the sequences, it's interesting to answer the question: why gene or protein sequences may be similar? It is indeed veryinteresting because the answer is related
-
5.4. The UPGMA algorithm
RechenmannFrançoisWe know how to fill an array with the values of the distances between sequences, pairs of sequences which are available in the file. This array of distances will be the input of our algorithm for








Sur le même thème
-
La voix, une donnée identifiante à protéger
VincentEmmanuelEmmanuel Vincent, chercheur au Centre Inria de l'Université de Lorraine et au Loria (Laboratoire lorrain de recherche en informatique et ses applications), présente sa recherche sur l'anonymisation de
-
Podcast 1/4 d'heure avec : Emmanuel Vincent, chercheur au Centre Inria de l'Université de Lorraine …
VincentEmmanuelRencontre avec Emmanuel Vincent - chercheur au Centre Inria de l'Université de Lorraine et Loria (Laboratoire lorrain de recherche en informatique et ses applications).
-
Stockage de données numériques sur ADN synthétique : Introduction au domaine
AntoniniMarcDuprazElsaLavenierDominiquePrésentation globale des différentes étapes du stockage de données sur des molécules d'ADN synthétique
-
Stockage de données numériques sur ADN synthétique : Production des données: synthèse, séquençage
LavenierDominiqueBarbryPascalDescription des opérations d'écriture et de lecture des molécules d'ADN : synthèse et séquençage.
-
Stockage de données numériques sur ADN synthétique : Reconstruction des données
LavenierDominiqueTraitement des données après séquençage
-
Stockage de données numériques sur ADN synthétique : Codage Canal
DuprazElsaTechniques de codage pour le stockage de données sur ADN
-
Stockage de données numériques sur ADN synthétique : Codage Source
AntoniniMarcCodage source pour le stockage de données sur ADN synthétique
-
Stockage de données numériques sur ADN synthétique : Théorie de l'information
Kas HannaSergeQuelle quantité d'information peut-on stocker et récupérer de manière fiable dans l'ADN ?
-
The tree of life
AbbySophieLes Rencontres Exobiologiques pour Doctorants (RED) sont une école de formation sur les « bases de l'astrobiologie ». L’édition 2025 s’est tenue du 16 au 21 mars au Parc Ornithologique du Teich.
-
Machines algorithmiques, mythes et réalités
MazenodVincentVincent Mazenod, informaticien, partage le fruit de ses réflexions sur l'évolution des outils numériques, en lien avec les problématiques de souveraineté, de sécurité et de vie privée...
-
Désassemblons le numérique - #Episode11 : Les algorithmes façonnent-ils notre société ?
SchwartzArnaudLima PillaLaércioEstériePierreSalletFrédéricFerbosAudeRoumanosRayyaChraibi KadoudIkramUn an après le tout premier hackathon sur les méthodologies d'enquêtes journalistiques sur les algorithmes, ce nouvel épisode part à la rencontre de différents points de vue sur les algorithmes.
-
Les machines à enseigner. Du livre à l'IA...
BruillardÉricQue peut-on, que doit-on déléguer à des machines ? C'est l'une des questions explorées par Éric Bruillard qui, du livre aux IA génératives, expose l'évolution des machines à enseigner...











